Numerous cell sources are being explored to replenish functional β-cell mass since the proof-of -concept for cell therapy of diabetes was laid down by transplantation of islets. Here, we explore vVarious strategies that aim to generate bone fide insulin producing cells. We focus i are explored. In particular on reprogramming and especially on α-cells conversion into insulin producing cells are focused here. A logical place to begin with for generating β-cells is to utilise the plasticity of closely related endoderm derived cell types like pancreatic non-β-cells and coaxing them to adopt a β-cell phenotype. Given the close ontogenetic relationship, functional similarity and dependency among these cells, the potential for interconversion is unequivocal. Phenotypic plasticity between pancreatic α-cells and β-cells is notably pronounced.
- β-cell
- α-cell
- diabetes mellitus
- cell sources
- reprogramming
- insulin
- transcription factors
- small molecules
1. Introduction
2. Reprogramming Cells to Make Insulin
For the last 20 years, significant efforts have focused on the generation of human embryonic stem cells, hESC-derived β-cells, that exhibit sustainable insulin secretion features, which mainly mirror the dynamicity of native human islets. By replicating the signaling events and physiologically relevant cues that dictate fate transition through definitive endoderm and pancreatic endoderm to hormone-expressing cells during human pancreas development, β-like cells can be produced [12,13,14][12][13][14]. The particular interest in hESCs is supported by the fact that these cells have the capacity of extensive self-renewal and virtually can be differentiated into derivatives of all three germ layers. Numerous studies have outlined differentiation protocols of insulin-producing cell types from hESCs [15,16,17,18,19,20,21][15][16][17][18][19][20][21]. However, reconstructing functional equivalent, mono-hormonal cells producing insulin under cell culture conditions has been intangible. This is likely because the in vitro systems utilized lack critical signals present in vivo, including the close interaction between exocrine, ductal and endocrine cells [15,20,22,23][15][20][22][23]. Among the critical concerns with hESC-derived therapeutic products is the occurrence of unwanted cell populations during in vitro differentiation that might interfere with the activity of the desired cell populations. Another concern is the risk of tumorigenicity. Moreover, the wider use of ESCs for research reasons is still largely hampered by numerous governmental bans concerning the use of human embryos in many countries, as well as by ethical and religious sensitivities. Compared to ESC and induced pluripotent stem cells (iPSCs), adult stem cell-like, mesenchymal stem cells (MSC) are considered ideal candidates to generate functional beta cells for personalized medicine owing to their superior anti-inflammatory, immunomodulatory and angiogenic properties. Numerous studies have shown that MSCs isolated from ample tissues and organs, such as bone marrow, adipose tissue, Wharton’s jelly, umbilical cords matrix blood, placenta and dental pulps, possess a developmental plasticity to differentiate into functional insulin producing cells with similar cytoarchitecture and functionality to β-cells. Consequently, utilizing MSC transplantation for the treatment of diabetes has been the focus of randomized controlled trails (RCTs) for the last few years. Although several clinical trials have shown that MSCs can reduce hyperglycemia by increasing insulin secretion in humans, the lack of control arms in some small sample sizes, inconsistent methods of isolation and delivery of MSCs, adverse effects and the failure to sustain therapeutic effect longitudinally from MSC therapy were common limitations in almost all RCTs. Finally, genetically modified animals designed for xenotransplantation or interspecies chimera-derived human organs, by using the blastocyst complementation method, could potentially offer unlimited sources of β-cells. Figure 1 summarizes several strategies aimed at generation of bone fide β-cell for replacement in diabetes.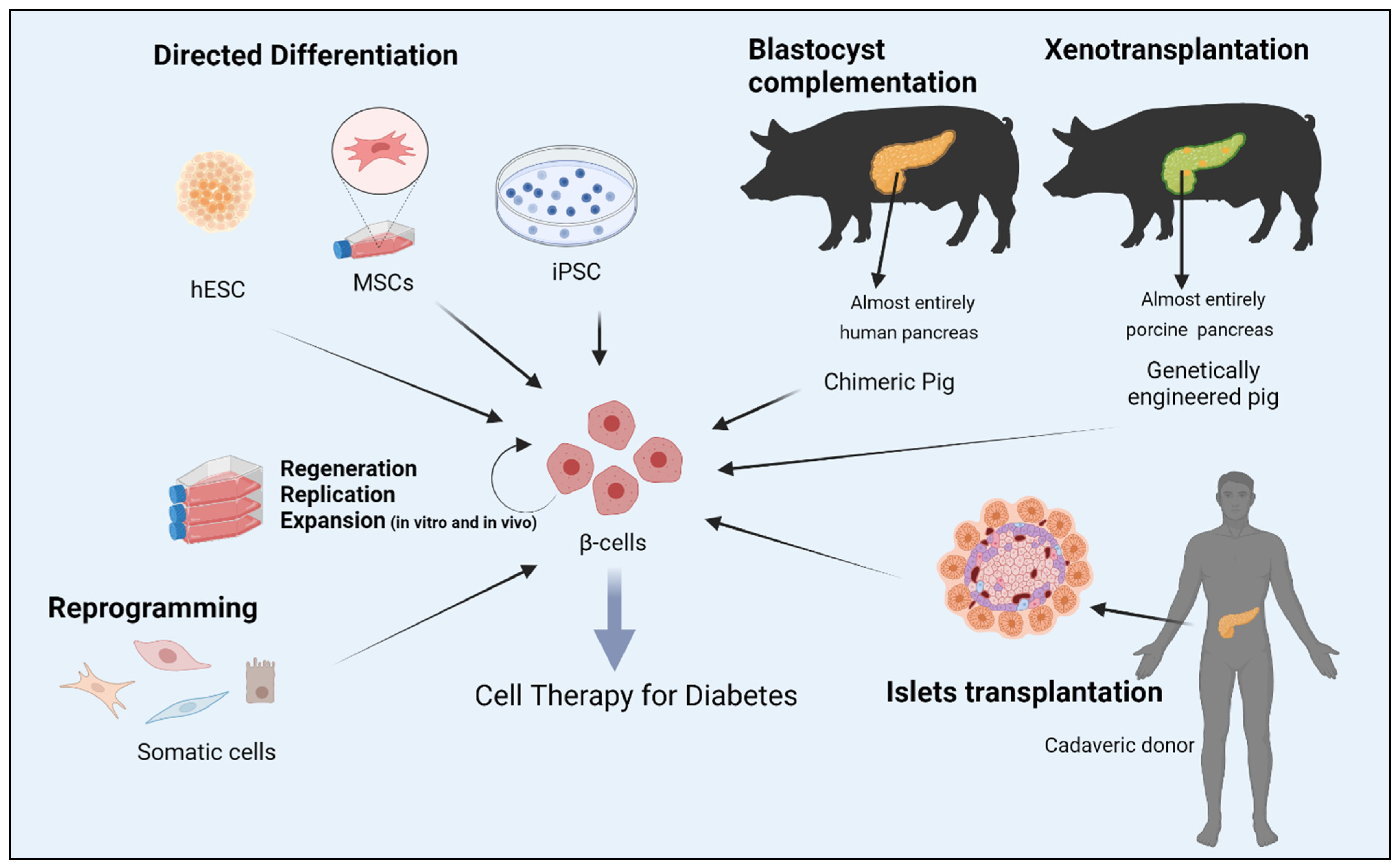
3. Alpha to β-cell Reprogramming
A logical place to begin with for generating β-cells is to utilise the plasticity of closely related endoderm derived cell types like pancreatic non-β-cells and coaxing them to adopt a β-cell phenotype. Given the close ontogenetic relationship, functional similarity and dependency among these cells, the potential for interconversion is unequivocal [24]. Phenotypic plasticity between pancreatic α-cells and β-cells is notably pronounced. A prominent study several years ago demonstrated that inter-endocrine plasticity and proliferation of β-cells can be elicited upon increased metabolic demand or after substantial diphtherial toxin induced β-cell loss [25]. While this sentudry was performed in mice, there is no evidence to suggest that a similar intrinsic cell type interconversion event can occur in other settings of β-cell damage in humans. Of note, a similar phenomenon of β-cell loss may naturally occur in mice transitioning into adulthood from puberty. Forced overexpression of key transcriptional regulators, the paired/homeodomain transcription factor (PAX4) [26], Insulin promoter factor 1 (PDX1) [27], or PDX1 and MAF BZIP Transcription Factor A (MAFA) [28], inhibition of DNA (cytosine-5)-methyltransferase 1 (DNMT1) and Aristaless related homeobox (ARX) [29], have also been shown to successfully drive α- to β-cell conversion. Yang et al. illustrated that PDX1 alone is not sufficient to allow the full reprogramming of glucagon-expressing cells [27]. This points to a chronotypic effect, where a subset of endocrine progenitors is primed for fate switch at the peri/postnatal stage, highlighting the role of the epigenome in reprogramming events. Importantly, the applicability of these findings to human cells needs to be determined, given that most of these studies were conducted in rodents. A subsequent study sought to better study human islet cell plasticity by lineage tracing and reprogramming α-cells with Mafa and Pdx1. Remarkably, insulin-producing α-cells retained α-cell markers, as evidenced by transcriptomic and proteomic characterization, while retaining insulin production and reversing diabetes for 6 months [30]. Several studies highlighted the type of pancreatic gene delivery systems for efficient intrinsic alpha cell transduction. Using adeno-associated virus (AAV) rather than adenoviral or lentiviral vectors has been shown to exhibit long-term gene expression induced reprogramming of pancreatic α-cells into functional β-cells [31]. The development of mature pancreatic endocrine cell subtypes represents the culmination of complex fate determining transcriptional programs that orchestrate the transition from one progenitor state to another. As transcription factors are sufficient to induce α- to β-cell reprogramming, it will be imperative to identify molecules that govern such a conversion as they would permit better control of the process [32]. For example, α-cells treated with glucagon-like peptide receptor agonists, exendin-4, can enhance pancreatic α-cells proliferation and their trans-differentiation into β-cells [33]. Similarly, sustained γ-Aminobutyric acid (GABA) exposure can induce concomitant α-cell to β-like cell neogenesis in vivo from human islet α-cells transplanted in mice by mobilization of duct-lining precursor cells that adopt an α cell identity [34]. GABA is a critical paracrine signal released by β-cells that inhibits glucagon-release from α-cells [35]. Artemisinin, a class of antimalarial agents, has been shown to impair α-cell identity in immortalized rodent cell lines by enhancing GABA receptor signaling, and by functionally inhibiting the ARX transcription factor [36]. In contrast, other studies have found that inhibition of ARX after long-term treatment by artemether, a derivative of artemisinin, did not furnish any α- to β-cell trans-differentiation in primary mouse islets, suggesting that these regulators may not represent a viable route to a novel diabetes therapy [37,38][37][38]. Nonetheless, in support of glucagon inhibition as a mechanism of α- to β- cell transition, the monoclonal antibody antagonist of the glucagon receptor (Ab-4) has been shown to enhance the formation of functional β-cell mass and their conversion from α-cell precursors in a rodent model of T1DM [39].4. Conclusions and Future Perspectives
Despite the vast potential of recent advances in cellular reprogramming and trans-differentiation of insulin producing cells, many issues still need refining in the coming decades. These include the efficiency of the cell trans-differentiation protocols, their cost-effectiveness for large-scale differentiation and whether reprogrammed cells can sustain their new differentiation state, or whether they potentially return to their early fate. Furthermore, whether transdifferentiated β-cells can respond adequately to the multitude of physiological stimuli required to maintain metabolic equilibrium and upon increased metabolic demand. Lastly, are the reprogrammed cells able to evade the recurrent auto-immune attacks, particularly in the context of allogenic cell transplants, and might cell transplants possess increased oncogenic potential resulting from incomplete epigenetic conversion? Several technologies have been proposed to attend to the immune responses to and blood supply for transplants, including the use of physical shielding by encapsulating the reprogrammed cells in a device that allows for insulin and nutrients to efficiently cross through the membrane while blocking cells from trespassing. Other biological interventions include either modifying host immune system, using antibodies to block the immune reactions or gene editing of cells used for transplant. Furthermore, the safe implementation of viral reprogramming technologies to the human setting need to be further assessed. The small molecules approach for reprogramming are more intriguing than virus-based methods, but they are very complex to develop for the regulation of a number of transcription factors. Moreover, impeding to clinical translation of the reprogramming approach still, is the relatively limited conversion rate into functional bone fide β-cells and presence of unwanted cells in the final product A paradigm shift has been made toward understanding fundamental β-cell biology, development and reprogramming over the last 20 years. Although a plethora of cell sources have been suggested as a starting material en route to successful reprogramming, some cells may fail. Thus, it is difficult to speculate which cells will reach the finish line and lead to a replacement therapy for diabetes. In conclusion, wresearchers have witnessed a remarkable two decades of significant progress in the methods of creating cellular transplant for the cure of diabetes. Current clinical trials for T1DM are still using hESC-derived pancreatic progenitors as a surrogate for cadaveric material, but the debate over whether to transplant a more mature cell population more similar to human islets is still ongoing. Hepatic, gastrointestinal and pancreatic exocrine cells, which are derived from common endodermal progenitor cells, have the potential to take the lead as cell sources for the development of a therapeutic product. Identical developmental transcription mechanisms and regulatory networks, analogous chromatin landscapes, physical proximity to the injured pancreas and a minimal need for epigenomic rearrangement are all tempting to conclude that these functionally related cells may yield more robust reprogramming outcomes, and offers a unique promise for cell replacement therapies for diabetes.References
- International Diabetes Federation. Diabetes Facts & Figures; International Diabetes Federation: Brussels, Belgium, 2021; Available online: https://idf.org/aboutdiabetes/what-is-diabetes/facts-figures.html (accessed on 1 September 2022).
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790.
- Chen, H.; Chen, G.; Zheng, X.; Guo, Y. Contribution of specific diseases and injuries to changes in health adjusted life expectancy in 187 countries from 1990 to 2013: Retrospective observational study. BMJ 2019, 364, l969.
- Holman, R.R.; Paul, S.K.; Bethel, M.A.; Matthews, D.R.; Neil, H.A. 10-year follow-up of intensive glucose control in type 2 diabetes. N. Engl. J. Med. 2008, 359, 1577–1589.
- Diabetes Control and Complications Trial (DCCT); Epidemiology of Diabetes Interventions and Complications (EDIC) Research Group. Effect of intensive diabetes therapy on the progression of diabetic retinopathy in patients with type 1 diabetes: 18 years of follow-up in the DCCT/EDIC. Diabetes 2015, 64, 631–642.
- Zinman, B.; Genuth, S.; Nathan, D.M. The Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study: 30th Anniversary Presentations. Diabetes Care 2014, 37, 8.
- Polonsky, K.S. The Past 200 Years in Diabetes. N. Engl. J. Med. 2012, 367, 1332–1340.
- NICE-Sugar Study Investigators. Hypoglycemia and risk of death in critically ill patients. N. Engl. J. Med. 2012, 367, 1108–1118.
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300.
- Brown, S.A.; Kovatchev, B.P.; Raghinaru, D.; Lum, J.W.; Buckingham, B.A.; Kudva, Y.C.; Laffel, L.M.; Levy, C.J.; Pinsker, J.E.; Wadwa, R.P.; et al. Six-Month Randomized, Multicenter Trial of Closed-Loop Control in Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 1707–1717.
- Shapiro, A.M.J.; Ricordi, C.; Hering, B.J.; Auchincloss, H.; Lindblad, R.; Robertson, R.P.; Secchi, A.; Brendel, M.D.; Berney, T.; Brennan, D.C.; et al. International Trial of the Edmonton Protocol for Islet Transplantation. N. Engl. J. Med. 2006, 355, 1318–1330.
- Oliver-Krasinski, J.M.; Stoffers, D.A. On the origin of the beta cell. Genes Dev. 2008, 22, 1998–2021.
- Assady, S.; Maor, G.; Amit, M.; Itskovitz-Eldor, J.; Skorecki, K.L.; Tzukerman, M. Insulin Production by Human Embryonic Stem Cells. Diabetes 2001, 50, 1691–1697.
- D’Amour, K.A.; Agulnick, A.D.; Eliazer, S.; Kelly, O.G.; Kroon, E.; Baetge, E.E. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat. Biotechnol. 2005, 23, 1534–1541.
- D’Amour, K.A.; Bang, A.G.; Eliazer, S.; Kelly, O.G.; Agulnick, A.D.; Smart, N.G.; Moorman, M.A.; Kroon, E.; Carpenter, M.K.; Baetge, E.E. Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat. Biotechnol. 2006, 24, 1392–1401.
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452.
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.-I.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell Biol. 2019, 21, 263–274.
- Hogrebe, N.J.; Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat. Biotechnol. 2020, 38, 460–470.
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133.
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439.
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772.
- Naujok, O.; Burns, C.; Jones, P.M.; Lenzen, S. Insulin-producing surrogate beta-cells from embryonic stem cells: Are we there yet? Mol. Ther. 2011, 19, 1759–1768.
- Pagliuca, F.W.; Melton, D.A. How to make a functional beta-cell. Development 2013, 140, 2472–2483.
- Hamaguchi, K.; Utsunomiya, N.; Takaki, R.; Yoshimatsu, H.; Sakata, T. Cellular interaction between mouse pancreatic alpha-cell and beta-cell lines: Possible contact-dependent inhibition of insulin secretion. Exp. Biol. Med. 2003, 228, 1227–1233.
- Thorel, F.; Népote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154.
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009, 138, 449–462.
- Yang, Y.P.; Thorel, F.; Boyer, D.F.; Herrera, P.L.; Wright, C.V. Context-specific alpha- to-beta-cell reprogramming by forced Pdx1 expression. Genes Dev. 2011, 25, 1680–1685.
- Matsuoka, T.A.; Kawashima, S.; Miyatsuka, T.; Sasaki, S.; Shimo, N.; Katakami, N.; Kawamori, D.; Takebe, S.; Herrera, P.L.; Kaneto, H.; et al. Mafa Enables Pdx1 to Effectively Convert Pancreatic Islet Progenitors and Committed Islet alpha-Cells Into beta-Cells In Vivo. Diabetes 2017, 66, 1293–1300.
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet alpha Cells into beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634.
- Furuyama, K.; Chera, S.; Van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human alpha-cells. Nature 2019, 567, 43–48.
- Xiao, X.; Guo, P.; Shiota, C.; Zhang, T.; Coudriet, G.M.; Fischbach, S.; Prasadan, K.; Fusco, J.; Ramachandran, S.; Witkowski, P.; et al. Endogenous Reprogramming of Alpha Cells into Beta Cells, Induced by Viral Gene Therapy, Reverses Autoimmune Diabetes. Cell Stem Cell 2018, 22, 78–90.e4.
- Fomina-Yadlin, D.; Kubicek, S.; Walpita, D.; Dančik, V.; Hecksher-Sørensen, J.; Bittker, J.A.; Sharifnia, T.; Shamji, A.; Clemons, P.A.; Wagner, B.K.; et al. Small-molecule inducers of insulin expression in pancreatic α-cells. Proc. Natl. Acad. Sci. USA 2010, 107, 15099–15104.
- Lee, Y.S.; Lee, C.; Choung, J.S.; Jung, H.S.; Jun, H.S. Glucagon-Like Peptide 1 Increases beta-Cell Regeneration by Promoting alpha- to beta-Cell Transdifferentiation. Diabetes 2018, 67, 2601–2614.
- Ben-Othman, N.; Vieira, A.; Courtney, M.; Record, F.; Gjernes, E.; Avolio, F.; Hadzic, B.; Druelle, N.; Napolitano, T.; Navarro-Sanz, S.; et al. Long-Term GABA Administration Induces Alpha Cell-Mediated Beta-like Cell Neogenesis. Cell 2017, 168, 73–85.e11.
- Menegaz, D.; Hagan, D.W.; Almaça, J.; Cianciaruso, C.; Rodriguez-Diaz, R.; Molina, J.; Dolan, R.M.; Becker, M.W.; Schwalie, P.C.; Nano, R.; et al. Mechanism and effects of pulsatile GABA secretion from cytosolic pools in the human beta cell. Nat. Metab. 2019, 1, 1110–1126.
- Li, J.; Casteels, T.; Frogne, T.; Ingvorsen, C.; Honoré, C.; Courtney, M.; Huber, K.V.; Schmitner, N.; Kimmel, R.A.; Romanov, R.A.; et al. Artemisinins Target GABAA Receptor Signaling and Impair α Cell Identity. Cell 2016, 168, 86–100.e15.
- van der Meulen, T.L.S.; Noordeloos, E.; Donaldson, C.J.; Adams, M.W.; Noguchi, G.M.; Mawla, A.M.; Huising, M.O. Artemether Does Not Turn α Cells into β Cells. Cell Metab. 2018, 27, 218–225.e4.
- Ackermann, A.M.; Moss, N.G.; Kaestner, K.H. GABA and Artesunate Do Not Induce Pancreatic alpha-to-beta Cell Transdifferentiation In Vivo. Cell Metab. 2018, 28, 787–792.e3.
- Wang, M.Y.; Dean, E.D.; Quittner-Strom, E.; Zhu, Y.; Chowdhury, K.H.; Zhang, Z.; Zhao, S.; Li, N.; Ye, R.; Lee, Y.; et al. Glucagon blockade restores functional beta-cell mass in type 1 diabetic mice and enhances function of human islets. Proc. Natl. Acad. Sci. USA 2021, 118, e2022142118.