 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Thomas Böldicke | -- | 3076 | 2022-09-01 13:24:39 | | | |
| 2 | Catherine Yang | -2 word(s) | 3074 | 2022-09-02 03:26:50 | | | | |
| 3 | Catherine Yang | + 58 word(s) | 3132 | 2022-09-02 10:36:47 | | |
Video Upload Options
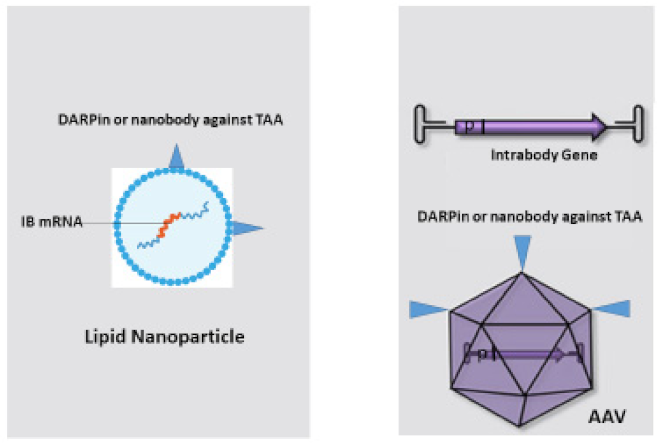
Tumor cells are characterized by overexpressed tumor-associated antigens or mutated neoantigens, which are expressed on the cell surface or intracellularly. One strategy of cancer immunotherapy is to target cell-surface-expressed tumor-associated antigens (TAAs) with therapeutic antibodies. Intrabodies are suitable to knockdown TAAs and neoantigens without off-target effects. Intrabodies can now be selected against virtually any protein inside the cell. RNA sequencing and proteome analysis of single tumor cells combined with computational methods is bringing forward the identification of new neoantigens for the selection of anti-cancer intrabodies, which can be easily performed using phage display antibody repertoires. Anti-cancer intrabodies demonstrated tumor growth inhibition in appropriate xenograft tumor mouse models. At the moment, the biggest challenge in translating TAA/neoantigen-directed intrabodies into the clinic is the specific targeting of the intrabodies to the tumor cells. The promising development of tumor-specific lipid nanoparticles which could be embedded with an mRNA transgene or new capsid-modified and tumor-specific recombinant AAVs should enable tumor-cell-specific intrabody transfection/transduction in cancer patients and may finally bring intrabodies into the clinic.
1. Introduction
Cancer immunotherapy with monoclonal antibodies or antibody fragments comprises the targeting of antibodies to extracellular or intracellular tumor-associated antigens (TAAs) or mutated neoantigens [1][2][3]. In contrast to TAAs, neoantigens comprise tumor-specific mutations and are not expressed by normal cells. The binding of tumor-specific antibodies to extracellular TAAs or neoantigens activates natural killer cells, macrophages or the complement system, leading to the destruction of the tumor cells. In addition to naked monoclonal antibodies, bispecific antibodies, immunotoxins, immunocytokines and engineered CAR-T cells with a TAA or neoantigen-specific antibody fragment or TCR-like antibody fused to T-cell signal domains can be successfully applied [4][5].
Intrabodies can now be selected against virtually any protein inside the cell and they have the potential to specifically inhibit the function of TAAs and even neoantigens in cancer patients. Two different kinds of intrabodies exist with different modes of action. One group of intrabodies comprises the ER intrabodies produced as scFvs inside the ER to inhibit the function of transitory proteins passing the secretory pathway [6]. Functional inhibition is performed through intrabody/antigen retention by the SE (KDEL) sequence fused to the C terminal end of the intrabody. Many ER intrabodies have been selected against overexpressed TAAs on the tumor cell surface [6][7][8].
The other group are single domain antibodies (sdAbs) comprising only the variable domain of the heavy chain VHH from camels (nanobodies) or sharks or human VH and VLs and are stable in the cytoplasm or nucleus [9]. They inactivate their targets by altering their conformation or interfere with the binding of the target protein to its corresponding binding partner.
Anti-cancer intrabodies demonstrated tumor growth inhibition in appropriate xenograft tumor mouse models [10][11][12][13][14][15][16][17][18][19][20]. Furthermore, a scFv-Fc intrabody inhibited the function of the serin (727)-phosphorylated form of STAT3 (pSSTAT3) in vitro and in mice [21]. STAT3 is involved in proliferation and apoptosis processes. CAR T cells transduced with an anti-CD7 intrabody applied in T-cell acute lymphoblastic leukemia (T-ALL) have been described [22]. The resulting CAR T cells only eliminated CD7+ lymphoblastic leukemia T cells and not the CAR T cells also expressing CD7.
2. Intrabodies against Oncogenic Cell Surface Receptors
3. Intrabodies against Cytoplasmic or Nucleus Located TAAS
4. Intrabodies against Intracellular Neoantigens
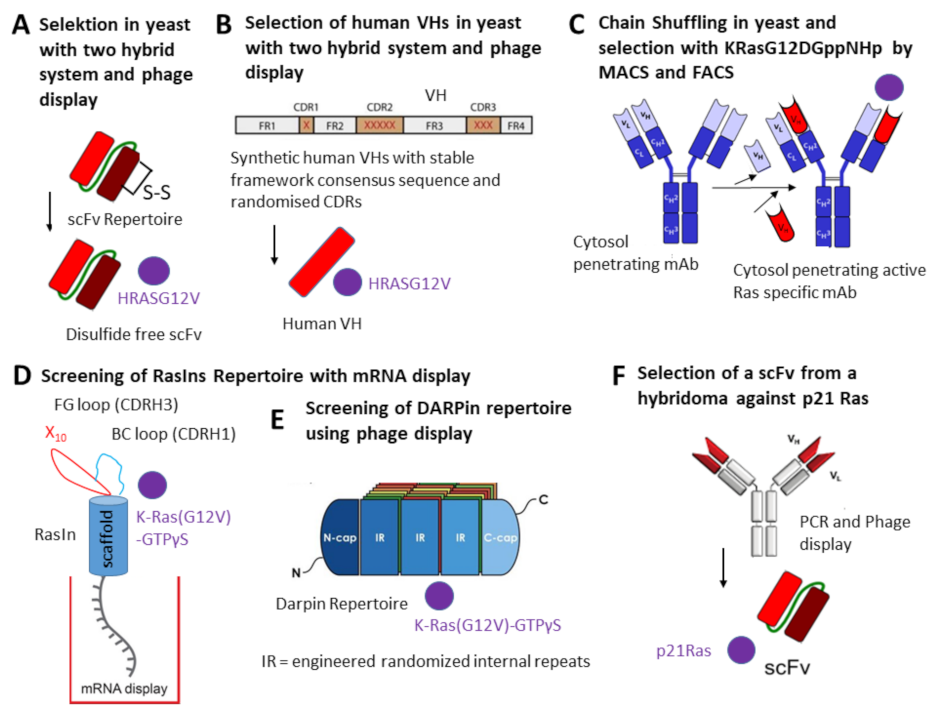
| Target | Selection of Intrabody | Physiological Knockdown Readout | Ref. |
|---|---|---|---|
| HRASG12V | A disulfide free scFv was selected with the intracellular antibody capture (IAC) technology [46]. | When NIH 3T3 cells were co-transfected with the disulfide-free scFv and RAS mutant-dependent luciferase construct the number of transformed foci was reduced to 30%. | [41] |
| HRASG12V | A human VH domain was selected from two synthetic VH domain libraries with fully randomized complementarity determining regions (CDRs) introduced into a single stable intrabody framework. One human VH was selected in yeast. | The VH binds to activated GTP-bound wild-type HRAS and HRAS (G12V). Nude mice were injected subcutaneously with mouse (NIH3T3-EJ) or human (HT-1080 or DLD-1) tumor cells transduced with the anti-Ras intrabody. Tumors were not developed in mice when the sdAb was expressed in the tumor cells compared to cells with an empty vector or an irrelevant intrabody. | [10] |
| GTP-bound K- and H-Ras and the corresponding G12V mutants | Antibody-like ligands as intrabodies were developed (RasIn1 and RasIn2). They were selected by mRNA display using an antigen K-Ras(G12V)-GTPγS. | Detailed binding analysis demonstrated that RasIn1 and RasIn2 recognized the binding domain of the Raf kinase in activated H-RasG12V. | [42] |
| Ras mutants | This anti-Ras mutant antibody was engineered from a human antibody previously generated and is internalized through clathrin-mediated endocytosis using heparan sulfate proteoglycan (HSPG) as a receptor and escapes from early endosomes into the cytosol [47]. | The intrabody (RT11-i) recognizes the GTP-bound active forms of wild-type (WT) KRas, NRas and HRas and their oncogenic mutants with mutations at positions 12, 13 or 61, such as KRasG12D, KRasG12V, KRasG13D, KRasQ61H, HRasG12V and NRasQ61R. RT11-i significantly inhibits the tumor growth of oncogenic Ras mutant tumor xenografts in mice. |
[11] |
| Ras mutants | A new human IgG intrabody (inRas37) binding to activated GTP-bound Ras mutants with two-fold stronger activity was engineered from RT11-i. | Inhibition of tumor growth was seen in several xenograft tumor mice bearing different preestablished colorectal tumors. | [12] |
| p21 Ras | scFv antibody was generated from a hybridoma. | The scFv recognizes wild-type H-p21Ras, K-p21Ras and N-p21Ras [12] and their mutated variants. The intrabody significantly inhibited the tumor growth of nude mice with established tumors derived from human colon cancer cell line SW480 or human liver cancer cell line BEL-7402. | [13] |
| KRASG12V | DARPins were selected from a phage display library by biopanning using biotinylated KRASG12V. | DARPins bound to an allosteric site of GDP or GTP-bound KRASWT and KRASG12V inhibiting KRAS nucleotide exchange and dimerization. | [43] |
5. Bringing Intrabodies into Cancer Patients: Delivery of Intrabodies with Nanoparticles or AAV
References
- Gerber, H.-P.; Sibener, L.V.; Lee, L.J.; Gee, M. Intracellular targets as source for cleaner targets for the treatment of solid tumors. Biochem. Pharmacol. 2019, 168, 275–284.
- Biernacki, M.A.; Bleakley, M. Neoantigens in Hematologic Malignancies. Front. Immunol. 2020, 11, 121.
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for Neoantigen Identification. Front. Immunol. 2019, 10, 1392.
- Naran, K.; Nundalall, T.; Chetty, S.; Barth, S. Principles of Immunotherapy: Implications for Treatment Strategies in Cancer and Infectious Diseases. Front. Microbiol. 2018, 9, 3158.
- Yarmarkovich, M.; Marshall, Q.F.; Warrington, J.M.; Premaratne, R.; Farrel, A.; Groff, D.; Li, W.; di Marco, M.; Runbeck, E.; Truong, H.; et al. Cross-HLA targeting of intracellular oncoproteins with peptide-centric CARs. Nature 2021, 599, 477–484.
- Marschall, A.L.; Dubel, S.; Boldicke, T. Recent Advances with ER Targeted Intrabodies. Adv. Exp. Med. Biol. 2016, 917, 77–93.
- Zhang, C.; Ötjengerdes, R.M.; Roewe, J.; Mejias-Estevez, R.; Marschall, A.L.J. Applying Antibodies Inside Cells: Principles and Recent Advances in Neurobiology, Virology and Oncology. BioDrugs 2020, 34, 435–462.
- Marschall, A.L.; Dubel, S.; Boldicke, T. Specific in vivo knockdown of protein function by intrabodies. MAbs 2015, 7, 1010–1035.
- Böldicke, T. Single domain antibodies for the knockdown of cytosolic and nuclear proteins. Protein Sci. 2017, 26, 925–945.
- Tanaka, T.; Williams, R.L.; Rabbitts, T.H. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. EMBO J. 2007, 26, 3250–3259.
- Shin, S.-M.; Choi, D.-K.; Bae, J.; Kim, J.-S.; Park, S.-W.; Kim, Y.-S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic admin-istration. Nat. Commun. 2017, 8, 15090.
- Shin, S.-M.; Kim, J.-S.; Park, S.-W.; Jun, S.-Y.; Kweon, H.-J.; Choi, D.-K.; Lee, D.; Cho, Y.B.; Kim, Y.-S. Direct targeting of oncogenic RAS mutants with a tumor-specific cytosol-penetrating antibody inhibits RAS mutant–driven tumor growth. Sci. Adv. 2020, 6, eaay2174.
- Yang, J.-L.; Pan, X.-Y.; Zhao, W.-X.; Hu, Q.-C.; Ding, F.; Feng, Q.; Li, G.-Y.; Luo, Y. The antitumor efficacy of a novel adenovirus-mediated anti-p21Ras single chain fragment variable antibody on human cancers in vitro and in vivo. Int. J. Oncol. 2016, 48, 1218–1228.
- Somplatzki, S.; Mühlenhoff, M.; Kröger, A.; Gerardy-Schahn, R.; Böldicke, T. Intrabodies against the Polysialyltransferases ST8SiaII and ST8SiaIV inhibit Polysialylation of NCAM in rhabdomyosarcoma tumor cells. BMC Biotechnol. 2017, 17, 42.
- Van Impe, K.; Bethuyne, J.; Cool, S.; Impens, F.; Ruano-Gallego, D.; De Wever, O.; Vanloo, B.; Van Troys, M.; Lambein, K.; Boucherie, C.; et al. A nanobody targeting the F-actin capping protein CapG restrains breast cancer metastasis. Breast Cancer Res. 2013, 15, R116.
- Deshane, J.; Siegal, G.P.; Wang, M.; Wright, M.; Bucy, R.P.; Alvarez, R.D.; Curiel, D.T. Transductional efficacy and safety of an intraperitoneally delivered adenovirus encoding an anti-erbB-2 intracellular single-chain antibody for ovarian cancer gene therapy. Gynecol. Oncol. 1997, 64, 378–385.
- Popkov, M.; Jendreyko, N.; McGavern, D.B.; Rader, C.; Barbas, C.F., 3rd. Targeting tumor angiogenesis with adenovi-rus-delivered anti-Tie-2 intrabody. Cancer Res. 2005, 65, 972–981.
- Jendreyko, N.; Popkov, M.; Rader, C.; Barbas, C.F. Phenotypic knockout of VEGF-R2 and Tie-2 with an intradiabody reduces tumor growth and angiogenesis in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 8293–8298.
- Chen, J.; Guo, H.; Jiang, H.; Namusamba, M.; Wang, C.; Lan, T.; Wang, T.; Wang, B. A BAP31 intrabody induces gastric cancer cell death by inhibiting p27(kip1) proteasome degradation. Int. J. Cancer 2019, 144, 2051–2062.
- Zhang, W.; Shan, H.; Jiang, K.; Huang, W.; Li, S. A novel intracellular nanobody against HPV16 E6 oncoprotein. Clin. Immunol. 2021, 225, 108684.
- Koo, M.Y.; Park, J.; Lim, J.M.; Joo, S.Y.; Shin, S.-P.; Shim, H.B.; Chung, J.; Kang, D.; Woo, H.A.; Rhee, S.G. Selective inhibition of the function of tyrosine-phosphorylated STAT3 with a phosphorylation site-specific intrabody. Proc. Natl. Acad. Sci. USA 2014, 111, 6269–6274.
- Png, Y.T.; Vinanica, N.; Kamiya, T.; Shimasaki, N.; Coustan-Smith, E.; Campana, D. Blockade of CD7 expression in T cells for effective chimeric antigen receptor targeting of T-cell malignancies. Blood Adv. 2017, 1, 2348–2360.
- Asaadi, Y.; Jouneghani, F.F.; Janani, S.; Rahbarizadeh, F. A comprehensive comparison between camelid nanobodies and single chain variable fragments. Biomark. Res. 2021, 9, 87.
- Böldicke, T.; Somplatzki, S.; Sergeev, G.; Mueller, P.P. Functional inhibition of transitory proteins by intrabody-mediated retention in the endoplasmatic reticulum. Methods 2012, 56, 338–350.
- Boldicke, T. Blocking translocation of cell surface molecules from the ER to the cell surface by intracellular antibodies tar-geted to the ER. J. Cell Mol. Med. 2007, 11, 54–70.
- Kirschning, C.J.; Dreher, S.; Maaß, B.; Fichte, S.; Schade, J.; Köster, M.; Noack, A.; Lindenmaier, W.; Wagner, H.; Böldicke, T. Generation of anti-TLR2 intrabody mediating inhibition of macrophage surface TLR2 expression and TLR2-driven cell activation. BMC Biotechnol. 2010, 10, 31.
- Böldicke, T.; Tesar, M.; Griesel, C.; Rohde, M.; Gröne, H.J.; Waltenberger, J.; Kollet, O.; Lapidot, T.; Yayon, A.; Weich, H. Anti-VEGFR-2 scFvs for cell isolation. Single-chain antibodies recognizing the human vascular endothelial growth factor receptor-2 (VEGFR-2/flk-1) on the surface of primary endothelial cells and preselected CD34+ cells from cord blood. Stem Cells 2001, 19, 24–36.
- Paolini, F.; Amici, C.; Carosi, M.; Bonomo, C.; Di Bonito, P.; Venuti, A.; Accardi, L. Intrabodies targeting human papillomavirus 16 E6 and E7 oncoproteins for therapy of established HPV-associated tumors. J. Exp. Clin. Cancer Res. 2021, 40, 37.
- Grimmig, T.; Moench, R.; Kreckel, J.; Haack, S.; Rueckert, F.; Rehder, R.; Tripathi, S.; Ribas, C.; Chandraker, A.; Germer, C.T.; et al. Toll Like Receptor 2, 4, and 9 Signaling Promotes Autoregulative Tumor Cell Growth and VEGF/PDGF Expression in Human Pancreatic Cancer. Int. J. Mol. Sci. 2016, 17, 2060.
- Al-Saraireh, Y.M.J.; Sutherland, M.; Springett, B.R.; Freiberger, F.; Morais, G.R.; Loadman, P.M.; Errington, R.J.; Smith, P.J.; Fukuda, M.; Gerardy-Schahn, R.; et al. Pharmacological Inhibition of polysialyltransferase ST8SiaII Modulates Tumour Cell Migration. PLoS ONE 2013, 8, e73366.
- Reimer, E.; Somplatzki, S.; Zegenhagen, D.; Hänel, S.; Fels, A.; Bollhorst, T.; Hovest, L.G.; Bauer, S.; Kirschning, C.J.; Böldicke, T. Molecular cloning and characterization of a novel anti-TLR9 intrabody. Cell. Mol. Biol. Lett. 2013, 18, 433–446.
- Böldicke, T. Single domain antibodies for the knockdown of cytosolic and nuclear proteins. Protein Sci. 2017, 26, 925–945.
- Kang, E.A.G.; Hu, M.; Ren, H.; Wang, J.; Cheng, X.; Li, R.; Yuan, B.; Balan, Y.; Bai, Z.; Huang, H. VHH212 nanobody targeting the hypoxia-inducible factor 1α suppresses angiogenesis and potentiates gemcitabine therapy in pancreatic cancer in vivo. Cancer Biol. Med. 2021, 18, 772–787.
- Shah, V.; Sheppard, B.; Sears, R.; Alani, A.W. Hypoxia: Friend or Foe for drug delivery in Pancreatic Cancer. Cancer Lett. 2020, 492, 63–70.
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405.
- D’Agostino, S.; Mazzega, E.; Praček, K.; Piccinin, S.; Pivetta, F.; Armellin, M.; Fortuna, S.; Maestro, R.; de Marco, A. Interference of p53:Twist1 interaction through competing nanobodies. Int. J. Biol. Macromol. 2022, 194, 24–31.
- De Groof, T.W.M.; Bergkamp, N.D.; Heukers, R.; Giap, T.; Bebelman, M.P.; Haas, R.G.-D.; Piersma, S.R.; Jimenez, C.R.; Garcia, K.C.; Ploegh, H.L.; et al. Selective targeting of ligand-dependent and -independent signaling by GPCR conformation-specific anti-US28 intrabodies. Nat. Commun. 2021, 12, 4357.
- Valdés-Tresanco, M.S.; Molina-Zapata, A.; Pose, A.G.; Moreno, E. Structural Insights into the Design of Synthetic Nanobody Libraries. Molecules 2022, 27, 2198.
- English, H.; Hong, J.; Ho, M. Ancient species offers contemporary therapeutics: An update on shark VNAR single domain antibody sequences, phage libraries and potential clinical applications. Antib. Ther. 2020, 3, 1–9.
- Kashima, D.; Kageoka, M.; Kimura, Y.; Horikawa, M.; Miura, M.; Nakakido, M.; Tsumoto, K.; Nagamune, T.; Kawahara, M. A Novel Cell-Based Intracellular Protein-Protein Interaction Detection Platform (SOLIS) for Multimo-dality Screening. ACS Synth. Biol. 2021, 10, 990–999.
- Tanaka, T.; Rabbitts, T.H. Intrabodies based on intracellular capture frameworks that bind the RAS protein with high af-finity and impair oncogenic transformation. EMBO J. 2003, 22, 1025–1035.
- Cetin, M.; Evenson, W.E.; Gross, G.G.; Jalali-Yazdi, F.; Krieger, D.; Arnold, D.; Takahashi, T.T.; Roberts, R.W. RasIns: Genetically Encoded Intrabodies of Activated Ras Proteins. J. Mol. Biol. 2017, 429, 562–573.
- Bery, N.; Legg, S.; Debreczeni, J.; Breed, J.; Embrey, K.; Stubbs, C.; Kolasinska-Zwierz, P.; Barrett, N.; Marwood, R.; Watson, J.; et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 2019, 10, 2607.
- Milburn, M.V.; Tong, L.; Devos, A.M.; Brünger, A.; Yamaizumi, Z.; Nishimura, S.; Kim, S.-H. Molecular Switch for Signal Transduction: Structural Differences Between Active and Inactive Forms of Protooncogenic ras Proteins. Science 1990, 247, 939–945.
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467.
- Visintin, M.; Quondam, M.; Cattaneo, A. The intracellular antibody capture technology: Towards the high-throughput se-lection of functional intracellular antibodies for target validation. Methods 2004, 34, 200–214.
- Choi, D.-K.; Bae, J.; Shin, S.-M.; Shin, J.-Y.; Kim, S.; Kim, Y.-S. A general strategy for generating intact, full-length IgG antibodies that penetrate into the cytosol of living cells. mAbs 2014, 6, 1402–1414.
- Huang, C.C.; Liu, F.R.; Feng, Q.; Pan, X.Y.; Song, S.L.; Yang, J.L. RGD4C peptide mediates anti-p21Ras scFv entry into tumor cells and produces an inhibitory effect on the human colon cancer cell line SW480. BMC Cancer 2021, 21, 321.
- Kawe, M.; Forrer, P.; Amstutz, P.; Plückthun, A. Isolation of Intracellular Proteinase Inhibitors Derived from Designed Ankyrin Repeat Proteins by Genetic Screening. J. Biol. Chem. 2006, 281, 40252–40263.
- Burns, T.F.; Borghaei, H.; Ramalingam, S.S.; Mok, T.S.; Peters, S. Targeting KRAS-Mutant Non-Small-Cell Lung Cancer: One Mutation at a Time, with a Focus on KRAS G12C Mutations. J. Clin. Oncol. 2020, 38, 4208–4218.
- Tang, D.; Kroemer, G.; Kang, R. Oncogenic KRAS blockade therapy: Renewed enthusiasm and persistent challenges. Mol. Cancer 2021, 20, 128.
- Jamal-Hanjani, M.; Thanopoulou, E.; Peggs, K.S.; Quezada, S.A.; Swanton, C. Tumour heterogeneity and im-mune-modulation. Curr. Opin. Pharmacol. 2013, 13, 497–503.
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56.
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469.
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206.
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124.
- Guevara, M.L.; Persano, F.; Persano, S. Advances in Lipid Nanoparticles for mRNA-Based Cancer Immunotherapy. Front. Chem. 2020, 8, 589959.
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41.
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles—From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015.
- Slastnikova, T.A.; Ulasov, A.V.; Rosenkranz, A.A.; Sobolev, A.S. Targeted Intracellular Delivery of Antibodies: The State of the Art. Front. Pharmacol. 2018, 9, 1208.
- Hacker, U.T.; Bentler, M.; Kaniowska, D.; Morgan, M.; Büning, H. Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives. Cancers 2020, 12, 1889.
- Colón-Thillet, R.; Jerome, K.R.; Stone, D. Optimization of AAV vectors to target persistent viral reservoirs. Virol. J. 2021, 18, 85.

