 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Letizia Caminiti | -- | 2805 | 2022-08-31 17:37:31 | | | |
| 2 | Lindsay Dong | -1 word(s) | 2804 | 2022-09-01 04:22:17 | | |
Video Upload Options
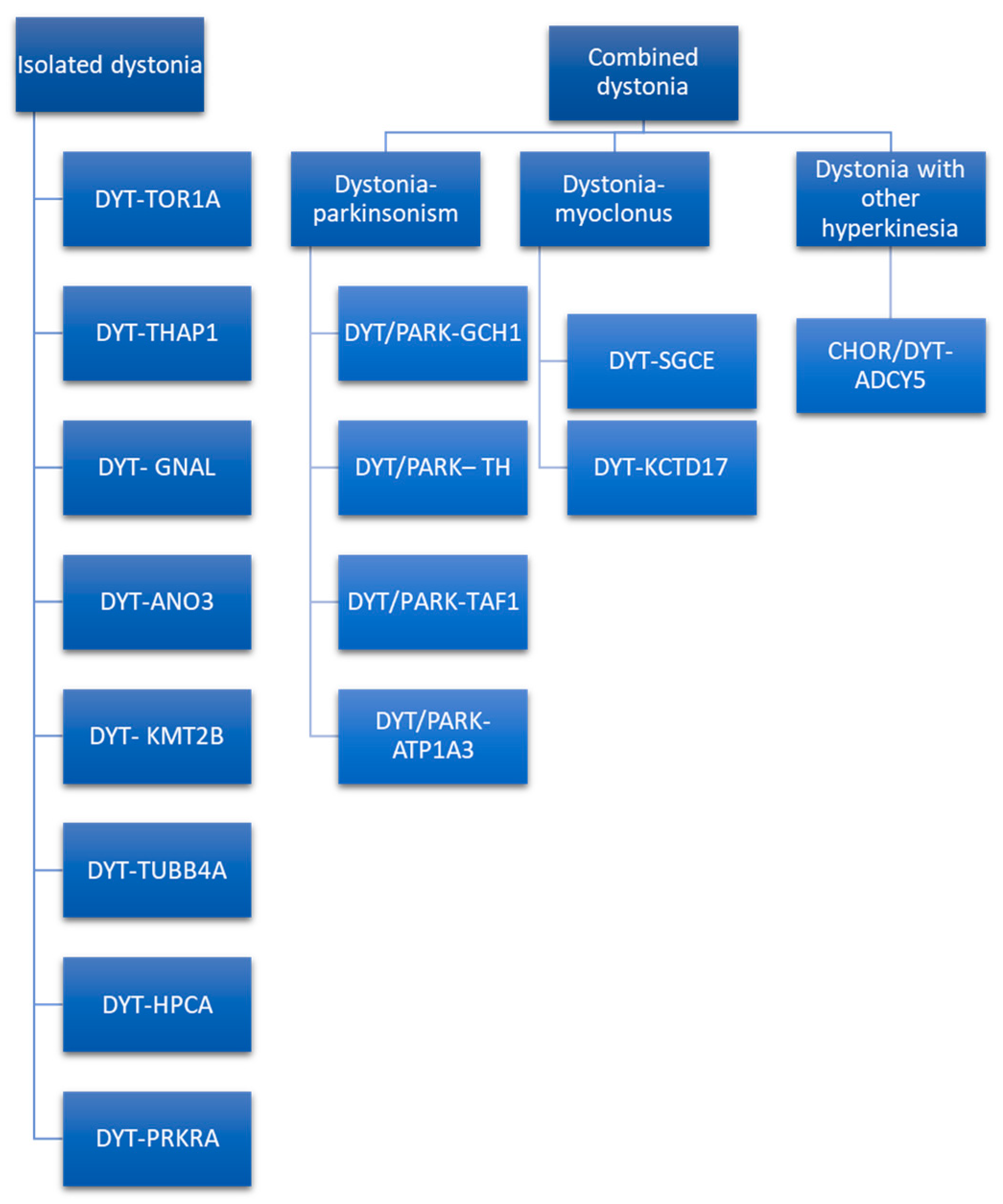
Dystonia diagnosis is based on clinical examination performed by a neurologist with expertise in movement disorders. Clues that indicate the diagnosis of a movement disorder such as dystonia are dystonic movements, dystonic postures, and three additional physical signs (mirror dystonia, overflow dystonia, and geste antagonists/sensory tricks). Despite advances in research, there is no diagnostic test with a high level of accuracy for the dystonia diagnosis. Clinical neurophysiology and genetics might support the clinician in the diagnostic process. Neurophysiology played a role in untangling dystonia pathophysiology, demonstrating characteristic reduction in inhibition of central motor circuits and alterations in the somatosensory system. The neurophysiologic measure with the greatest evidence in identifying patients affected by dystonia is the somatosensory temporal discrimination threshold (STDT). Other parameters need further confirmations and more solid evidence to be considered as support for the dystonia diagnosis. Genetic testing should be guided by characteristics such as age at onset, body distribution, associated features, and coexistence of other movement disorders (parkinsonism, myoclonus, and other hyperkinesia).
1. Introduction
-
Dystonia is a movement disorder characterized by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures, or both.
-
Dystonic movements are typically patterned, twisting, and may be tremulous.
-
Dystonia is often initiated or worsened by voluntary action and associated with overflow muscle activation.
2. Clinical Neurophysiology
3. Dystonia Genetics
References
- Albanese, A.; Bhatia, K.; Bressman, S.B.; DeLong, M.R.; Fahn, S.; Fung, V.S.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and classification of dystonia: A consensus update. Mov. Disord. 2013, 28, 863–873.
- Albanese, A.; Lalli, S. Is this dystonia? Mov. Disord. 2009, 24, 1725–1731.
- Albanese, A.; Di Giovanni, M.; Lalli, S. Dystonia: Diagnosis and management. Eur. J. Neurol. 2018, 26, 5–17.
- Di Biase, L.; Di Santo, A.; Caminiti, M.L.; Pecoraro, P.M.; Di Lazzaro, V. Classification of Dystonia. Life 2022, 12, 206.
- Van Gerpen, J.A.; Matsumoto, J.Y.; Ahlskog, J.E.; Maraganore, D.M.; McManis, P.G. Utility of an EMG mapping study in treating cervical dystonia. Muscle Nerve 2000, 23, 1752–1756.
- Edwards, M.J.; Talelli, P.; Rothwell, J.C. Clinical applications of transcranial magnetic stimulation in patients with movement disorders. Lancet Neurol. 2008, 7, 827–840.
- Chen, R.; Cros, D.; Curra, A.; Di Lazzaro, V.; Lefaucheur, J.-P.; Magistris, M.R.; Mills, K.; Rösler, K.M.; Triggs, W.J.; Ugawa, Y.; et al. The clinical diagnostic utility of transcranial magnetic stimulation: Report of an IFCN committee. Clin. Neurophysiol. 2008, 119, 504–532.
- Fregni, F.; Boggio, P.S.; Santos, M.C.; Lima, M.; Vieira, A.L.; Rigonatti, S.P.; Silva, M.T.A.; Barbosa, E.R.; Nitsche, M.A.; Pascual-Leone, A. Noninvasive cortical stimulation with transcranial direct current stimulation in Parkinson’s disease. Mov. Disord. 2006, 21, 1693–1702.
- Ferrucci, R.; Mameli, F.; Ruggiero, F.; Priori, A. Transcranial direct current stimulation as treatment for Parkinson’s disease and other movement disorders. Basal Ganglia 2015, 6, 53–61.
- Darrow, D.P. Focused Ultrasound for Neuromodulation. Neurotherapeutics 2019, 16, 88–99.
- Di Biase, L.; Falato, E.; Di Lazzaro, V. Transcranial Focused Ultrasound (tFUS) and Transcranial Unfocused Ultrasound (tUS) Neuromodulation: From Theoretical Principles to Stimulation Practices. Front. Neurol. 2019, 10, 549.
- Di Biase, L.; Falato, E.; Caminiti, M.L.; Pecoraro, P.M.; Narducci, F.; Di Lazzaro, V. Focused Ultrasound (FUS) for Chronic Pain Management: Approved and Potential Applications. Neurol. Res. Int. 2021, 2021, 1–16.
- Hallett, M. Chapter 1 Movement disorders: Overview. In Handbook of Clinical Neurophysiology; Elsevier: Amsterdam, The Netherlands, 2003; pp. 3–4.
- Bologna, M.; Suppa, A.; Di Stasio, F.; Conte, A.; Fabbrini, G.; Berardelli, A. Neurophysiological studies on atypical parkinsonian syndromes. Park. Relat. Disord. 2017, 42, 12–21.
- Valls-Solé, J. Neurophysiological characterization of parkinsonian syndromes. Neurophysiol. Clin. Neurophysiol. 2000, 30, 352–367.
- Valls-Solé, J.; Valldeoriola, F. Neurophysiological correlate of clinical signs in Parkinson’s disease. Clin. Neurophysiol. 2002, 113, 792–805.
- Di Biase, L.; Brittain, J.-S.; Shah, S.A.; Pedrosa, D.; Cagnan, H.; Mathy, A.; Chen, C.C.; Martín-Rodríguez, J.F.; Mir, P.; Timmerman, L.; et al. Tremor stability index: A new tool for differential diagnosis in tremor syndromes. Brain 2017, 140, 1977–1986.
- Di Pino, G.; Formica, D.; Melgari, J.M.; Taffoni, F.; Salomone, G.; di Biase, L.; Caimo, E.; Vernieri, F.; Guglielmelli, E. Neurophysiological bases of tremors and accelerometric parameters analysis. In Proceedings of the 2012 4th IEEE RAS & EMBS International Conference on Biomedical Robotics and Biomechatronics (BioRob), Rome, Italy, 24−27 June 2012; pp. 1820–1825.
- Deuschl, G.; Krack, P.; Lauk, M.; Timmer, J. Clinical Neurophysiology of Tremor. J. Clin. Neurophysiol. 1996, 13, 110–121.
- Caviness, J.N. Chapter 32 The clinical neurophysiology of myoclonus. In Handbook of Clinical Neurophysiology; Hallett, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 521–548.
- Kaji, R. Chapter 28 Dystonia. In Handbook of Clinical Neurophysiology; Hallett, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 451–461.
- Hallett, M. Neurophysiology of dystonia: The role of inhibition. Neurobiol. Dis. 2011, 42, 177–184.
- Conte, A.; Rocchi, L.; Latorre, A.; Belvisi, D.; Rothwell, J.C.; Berardelli, A. Ten-Year Reflections on the Neurophysiological Abnormalities of Focal Dystonias in Humans. Mov. Disord. 2019, 34, 1616–1628.
- Latorre, A.; Rocchi, L.; Bhatia, K.P. Delineating the electrophysiological signature of dystonia. Exp. Brain Res. 2020, 238, 1685–1692.
- Rothwell, J.C.; Obeso, J.A.; Day, B.L.; Marsden, C.D. Pathophysiology of dystonias. Adv. Neurol. 1983, 39, 851–863.
- Nakashima, K.; Rothwell, J.C.; Day, B.L.; Thompson, P.D.; Shannon, K.; Marsden, C.D. Reciprocal inhibition between forearm muscles in patients with writer’s cramp and other occupational cramps, symptomatic hemidystonia and hemiparesis due to stroke. Brain 1989, 112, 681–697.
- Berardelli, A.; Rothwell, J.; Day, B.L.; Marsden, C.D. Pathophysiology of blepharospasm and oromandibular dystonia. Brain 1985, 108, 593–608.
- Quartarone, A.; Girlanda, P.; Di Lazzaro, V.; Majorana, G.; Battaglia, F.; Messina, C. Short latency trigemi-no-sternocleidomastoid response in muscles in patients with spasmodic torticollis and blepharospasm. Clin. Neurophysiol. 2000, 111, 1672–1677.
- Huang, Y.-Z.; Rothwell, J.; Lu, C.-S.; Wang, J.-J.; Chen, R.-S. Restoration of motor inhibition through an abnormal premotor-motor connection in dystonia. Mov. Disord. 2010, 25, 696–703.
- Espay, A.; Morgante, F.; Purzner, J.; Gunraj, C.A.; Lang, A.; Chen, R. Cortical and spinal abnormalities in psychogenic dystonia. Ann. Neurol. 2006, 59, 825–834.
- Di Lazzaro, V.; Oliviero, A.; Profice, P.; Dileone, M.; Pilato, F.; Insola, A.; Della Marca, G.; Tonali, P.; Mazzone, P. Reduced cerebral cortex inhibition in dystonia: Direct evidence in humans. Clin. Neurophysiol. 2009, 120, 834–839.
- Beck, S.; Shamim, E.A.; Richardson, S.P.; Schubert, M.; Hallett, M. Inter-hemispheric inhibition is impaired in mirror dystonia. Eur. J. Neurosci. 2009, 29, 1634–1640.
- Chen, R.; Wassermann, E.M.; Caños, M.; Hallett, M. Impaired inhibition in writer’s cramp during voluntary muscle activation. Neurology 1997, 49, 1054–1059.
- Tinazzi, M.; Farina, S.; Edwards, M.; Moretto, G.; Restivo, D.; Fiaschi, A.; Berardelli, A. Task−specific impairment of motor cortical excitation and inhibition in patients with writer’s cramp. Neurosci. Lett. 2005, 378, 55–58.
- Bologna, M.; Berardelli, A. The cerebellum and dystonia. Handb. Clin. Neurol. 2018, 155, 259–272.
- Kojovic, M.; Pareés, I.; Kassavetis, P.; Palomar, F.J.; Mir, P.; Teo, J.; Cordivari, C.; Rothwell, J.; Bhatia, K.; Edwards, M.J. Secondary and primary dystonia: Pathophysiological differences. Brain 2013, 136, 2038–2049.
- Sadnicka, A.; Teo, J.; Kojovic, M.; Pareés, I.; Saifee, T.A.; Kassavetis, P.; Schwingenschuh, P.; Katschnig−Winter, P.; Stamelou, M.; Mencacci, N.E.; et al. All in the blink of an eye: New insight into cerebellar and brainstem function in DYT1 and DYT6 dystonia. Eur. J. Neurol. 2014, 22, 762–767.
- Conte, A.; Ferrazzano, G.; Belvisi, D.; Manzo, N.; Battista, E.; Voti, P.L.; Nardella, A.; Fabbrini, G.; Berardelli, A. Somatosensory temporal discrimination in Parkinson’s disease, dystonia and essential tremor: Pathophysiological and clinical implications. Clin. Neurophysiol. 2018, 129, 1849–1853.
- Quartarone, A.; Bagnato, S.; Rizzo, V.; Siebner, H.R.; Dattola, V.; Scalfari, A.; Morgante, F.; Battaglia, F.; Romano, M.; Girlanda, P. Abnormal associative plasticity of the human motor cortex in writer’s cramp. Brain 2003, 126, 2586–2596.
- Edwards, M.J.; Huang, Y.-Z.; Mir, P.; Rothwell, J.; Bhatia, K.P. Abnormalities in motor cortical plasticity differentiate manifesting and nonmanifesting DYT1 carriers. Mov. Disord. 2006, 21, 2181–2186.
- Quartarone, A.; Rizzo, V.; Terranova, C.; Morgante, F.; Schneider, S.; Ibrahim, N.; Girlanda, P.; Bhatia, K.P.; Rothwell, J.C. Abnormal sensorimotor plasticity in organic but not in psychogenic dystonia. Brain 2009, 132, 2871–2877.
- Dileone, M.; Profice, P.; Pilato, F.; Alfieri, P.; Cesarini, L.; Mercuri, E.; Leoni, C.; Tartaglia, M.; Di Iorio, R.; Zampino, G.; et al. Enhanced human brain associative plasticity in Costello syndrome. J. Physiol. 2010, 588, 3445–3456.
- Dileone, M.; Zampino, G.; Profice, P.; Pilato, F.; Leoni, C.; Ranieri, F.; Capone, F.; Tartaglia, M.; Brown, P.; Di Lazzaro, V. Dystonia in Costello syndrome. Park. Relat. Disord. 2012, 18, 798–800.
- Piña-Fuentes, D.; Beudel, M.; Little, S.; van Zijl, J.; Elting, J.W.; Oterdoom, D.L.M.; van Egmond, M.E.; van Dijk, J.M.C.; Tijssen, M.A.J. Toward adaptive deep brain stimulation for dystonia. Neurosurg. Focus 2018, 45, E3.
- Assenza, G.; Capone, F.; di Biase, L.; Ferreri, F.; Florio, L.; Guerra, A.; Marano, M.; Paolucci, M.; Ranieri, F.; Salomone, G. Oscillatory activities in neurological disorders of elderly: Biomarkers to target for neuromodulation. Front. Aging Neurosci. 2017, 9, 189.
- Silberstein, P.; Kühn, A.A.; Kupsch, A.; Trottenberg, T.; Krauss, J.K.; Wöhrle, J.C.; Mazzone, P.; Insola, A.; Di Lazzaro, V.; Oliviero, A.; et al. Patterning of globus pallidus local field potentials differs between Parkinson’s disease and dystonia. Brain 2003, 126, 2597–2608.
- Chen, C.C.; Kühn, A.A.; Trottenberg, T.; Kupsch, A.; Schneider, G.-H.; Brown, P. Neuronal activity in globus pallidus interna can be synchronized to local field potential activity over 3–12 Hz in patients with dystonia. Exp. Neurol. 2006, 202, 480–486.
- Starr, P.A.; Rau, G.M.; Davis, V.; Marks, W.J.; Ostrem, J.L.; Simmons, D.; Lindsey, N.; Turner, R. Spontaneous Pallidal Neuronal Activity in Human Dystonia: Comparison With Parkinson’s Disease and Normal Macaque. J. Neurophysiol. 2005, 93, 3165–3176.
- Vitek, J.L.; Delong, M.R.; Starr, P.A.; Hariz, M.I.; Metman, L.V. Intraoperative neurophysiology in DBS for dystonia. Mov. Disord. 2011, 26, S31–S36.
- Bressman, S.B. Dystonia genotypes, phenotypes, and classification. Adv. Neurol. 2004, 94, 101–107.
- Camargos, S.; Cardoso, F. Understanding dystonia: Diagnostic issues and how to overcome them. Arq. Neuropsiquiatr. 2016, 74, 921–936.
- Kramer, P.L.; De Leon, D.; Ozelius, L.; Risch, N.; Bressman, S.B.; Brin, M.F.; Schuback, D.E.; Burke, R.E.; Kwiatkowski, D.J.; Shale, H.; et al. Dystonia gene in Ashkenazi Jewish population is located on chromosome 9q32-34. Ann. Neurol. 1990, 27, 114–120.
- Marras, C.; Lohmann, K.; Lang, A.; Klein, C. Fixing the broken system of genetic locus symbols: Parkinson disease and dystonia as examples. Neurology 2012, 78, 1016–1024.
- Marras, C.; Lang, A.; Van De Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457.
- Lange, L.M.; Junker, J.; Loens, S.; Baumann, H.; Olschewski, L.; Schaake, S.; Madoev, H.; Petkovic, S.; Kuhnke, N.; Kasten, M.; et al. Genotype–Phenotype Relations for Isolated Dystonia Genes: MDSGene Systematic Review. Mov. Disord. 2021, 36, 1086–1103.
- Weissbach, A.; Saranza, G.; Domingo, A. Combined dystonias: Clinical and genetic updates. J. Neural Transm. 2020, 128, 417–429.
- Risch, N.J.; Bressman, S.B.; Senthil, G.; Ozelius, L.J. Intragenic Cis and Trans Modification of Genetic Susceptibility in DYT1 Torsion Dystonia. Am. J. Hum. Genet. 2007, 80, 1188–1193.
- Djarmati, A.; A Schneider, S.; Lohmann, K.; Winkler, S.; Pawlack, H.; Hagenah, J.; Brüggemann, N.; Zittel, S.; Fuchs, T.; Raković, A.; et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: A genetic screening study. Lancet Neurol. 2009, 8, 447–452.
- Hersheson, J.; Mencacci, N.E.; Davis, M.; Macdonald, N.; Trabzuni, D.; Ryten, M.; Pittman, A.; Paudel, R.; Kara, E.; Fawcett, K.; et al. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann. Neurol. 2012, 73, 546–553.
- Wilcox, R.A.; Winkler, S.; Lohmann, K.; Klein, C. Whispering dysphonia in an Australian family (DYT4): A clinical and genetic reappraisal. Mov. Disord. 2011, 26, 2404–2408.
- Ichinose, H.; Ohye, T.; Takahashi, E.-I.; Seki, N.; Hori, T.-A.; Segawa, M.; Nomura, Y.; Endo, K.; Tanaka, H.; Tsuji, S.; et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat. Genet. 1994, 8, 236–242.
- Makino, S.; Kaji, R.; Ando, S.; Tomizawa, M.; Yasuno, K.; Goto, S.; Matsumoto, S.; Tabuena, M.D.; Maranon, E.; Dantes, M.; et al. Reduced Neuron-Specific Expression of the TAF1 Gene Is Associated with X-Linked Dystonia-Parkinsonism. Am. J. Hum. Genet. 2007, 80, 393–406.
- Lee, L.V.; Pascasio, F.M.; Fuentes, F.D.; Viterbo, G.H. Torsion dystonia in Panay, Philippines. Adv. Neurol. 1976, 14, 137–151.
- Brüggemann, N.; Heldmann, M.; Klein, C.; Domingo, A.; Rasche, D.; Tronnier, V.; Rosales, R.L.; Jamora, R.D.G.; Lee, L.V.; Münte, T.F. Neuroanatomical changes extend beyond striatal atrophy in X-linked dystonia parkinsonism. Park. Relat. Disord. 2016, 31, 91–97.
- Song, P.C.; Le Bs, H.; Acuna, P.; De Guzman, J.K.P.; Sharma, N.; Ba, T.N.F.; Dy, M.E.; Go, C.L. Voice and swallowing dysfunction in X-linked dystonia parkinsonism. Laryngoscope 2019, 130, 171–177.
- Mencacci, N.E.; Rubio-Agusti, I.; Zdebik, A.; Asmus, F.; Ludtmann, M.H.; Ryten, M.; Plagnol, V.; Hauser, A.-K.; Bandres-Ciga, S.; Bettencourt, C.; et al. A Missense Mutation in KCTD17 Causes Autosomal Dominant Myoclonus−Dystonia. Am. J. Hum. Genet. 2015, 96, 938–947.
- Ferrini, A.; Steel, D.; Barwick, K.; Kurian, M.A. An Update on the Phenotype, Genotype and Neurobiology of ADCY5-Related Disease. Mov. Disord. 2021, 36, 1104–1114.
- Méneret, A.; Gras, D.; McGovern, E.; Roze, E. Caffeine and the Dyskinesia Related to Mutations in the ADCY5 Gene. Ann. Intern. Med. 2019, 171, 439.
- Artusi, C.A.; Dwivedi, A.; Romagnolo, A.; Bortolani, S.; Marsili, L.; Imbalzano, G.; Sturchio, A.; Keeling, E.G.; Zibetti, M.; Contarino, M.F.; et al. Differential response to pallidal deep brain stimulation among monogenic dystonias: Systematic review and meta−analysis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 426–433.

