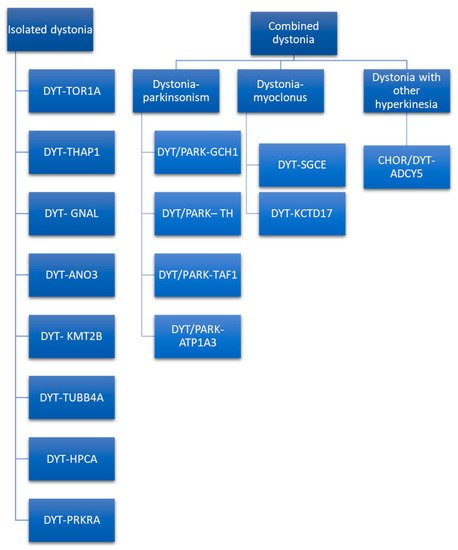
Moreover, in the proposed nomenclature and in the last consensus update on dystonia, the term complex dystonia is used, referring to conditions in which dystonia predominates the clinical phenotype but occurs in the context of a complex disease including symptoms other than movement disorders [
1,
69]. For example, Wilson disease is named according to the proposed nomenclature with a DYT prefix (DYT-ATP7B), and the same happens for Lesch–Nyhan syndrome and other infantile and childhood onset disease [
69]. Given that most of isolated hereditary dystonia is recognized as an autosomal dominant inheritance, the mode of transmission cannot be used as the only criterion to make a differential diagnosis. To guide the clinician towards a genetic diagnosis of dystonia, at least clinical phenotype and age of onset should be considered. If dystonia dominates the clinical picture, one of the isolated dystonias may be considered, and the gene mutations involved may be DYT-TOR1A, DYT-THAP1, DYT-GNAL, DYT-ANO3, DYT-KMT2B, DYT-TUBB4A, DYT-HPCA, and DYT-PRKRA [
70]. The last-mentioned dystonia is a controversial classification, as it is considered as combined dystonia by some authors [
71] and as isolated dystonia by others [
70]. Indeed, despite parkinsonism being described in about half the patients, it seemed to be caused not by true parkinsonian features, but by slow movements of dystonic body parts [
70]. The isolated form of dystonia could be distinguished according to the age of onset, body distribution, temporal pattern, associated features, responses to drugs, response to DBS, and brain imaging. Regarding age of onset, in infancy, childhood, and adolescence DYT-TOR1A, DYT-THAP1, DYT-KMT2B, DYT-TUBB4A, DYT-PRKRA, and DYT-HPCA are more probable, while DYT-ANO3 and DYT-GNAL begin in early adulthood. In particular, DYT-ANO3 recognizes two peaks of the age of onset: one in infancy/childhood and one in early-late adulthood [
70]. Age at onset may by modified by several aspects, e.g., penetrance as is the case of DYT-TOR1A [
72]. Hence, age of onset alone cannot be used as the only criteria to orient the diagnosis. According to body distribution, generalized forms of isolated dystonia are mainly due to DYT-TOR1A, DYT-THAP1, DYT-KMT2B, DYT-HPCA, and DYT-PRKRA. Among these, DYT-TOR1A, DYT-HPCA, and DYT-KMT2B usually begin in the lower limbs asymmetrically with secondary generalization. In contrast, DYT-THAP1 may initiate in the upper part of the body, involving cranio–cervical districts, speech difficulties, and the upper limbs, with successive generalizations [
73]. If DYT-TOR1A begins in the upper limbs, it tends to be focal. Focal and segmental isolated dystonia are more likely caused by DYT-GNAL and DYT-ANO3. These two forms of dystonia typically begin at the cervical level and may cause head tremor [
70]. DYT-GNAL may be suspected if age at onset is in early-late adulthood. In case of early involvement of craniofacial muscles with laryngeal dystonia and speech difficulties, with secondary generalization involving the arms at younger ages, DYT-ANO3 becomes more probable [
70]. Another peculiar form of isolated dystonia with focal distribution involving the cervical district and causing spasmodic dysphonia is caused by DYT-TUBB4A. This focal form may successively evolve into a generalized dystonia [
74]. Regarding the temporal pattern, except for the last-mentioned dystonia, all the other isolated dystonia follows a persistent temporal pattern. Associated features may guide the clinician in the differential diagnosis. The presence of additional phenotypic characteristic, such as microcephaly, short stature, intellectual disability, abnormal eye movements, myoclonus, dysmorphisms, and psychiatric symptoms, may be suggestive of DYT-KMT2B [
70]. Thin face, body habitus, and hobby horse gait are described in the DYT-TUBB4A [
75]. None of the isolated forms of dystonia respond to L-Dopa; DYT-TOR1A, DYT-THAP1, DYT-ANO3, DYT-KMT2B, and DYT-HPCA may respond to anticholinergics [
70]. Response to alcohol is described in DYT-GNAL and DYT-TUBB4A. It is important to define the genetic etiology of the dystonia because response to DBS varies according to the genetic conditions, and this is an important prognostic factor to be considered when selecting patients for advanced therapy.
Combined dystonia is characterized by the coexistence of another movement disorder in addition to dystonia. The association of dystonia with parkinsonism defines dystonia–parkinsonism. The monogenic forms of dystonia–parkinsonism are DYT/PARK-GCH1, DYT/PARK-TH, DYT/PARK-TAF1, and DYT/PARK-ATP1A3 [
71]. Contrary to what has been observed for isolated dystonia, combined dystonia recognizes a different mode of inheritance: autosomal dominant inheritance is characteristic of DYT/PARK-GCH1 and DYT/PARK-ATP1A3, while autosomal recessive inheritance is typical of DYT/PARK-TH. X-linked transmission characterizes DYT/PARK-TAF1 (also known as Lubag syndrome). Among this, it is of paramount importance to diagnose the dopa-responsive dystonia, DYT/PARK-GCH1. Indeed, patients have excellent and sustained response to L-Dopa [
80]. Another form of combined dystonia with response to L-Dopa is DYT/PARK-TH. These two forms of dystonia–parkinsonism may be differentiated according to age of onset, as DYT/PARK-GCH1 begins in infancy/childhood, while DYT/PARK-TH may initiate in infancy. Moreover, diurnal fluctuations of parkinsonian symptoms due to circadian variations in dopamine concentration are more pronounced in DYT/PARK-GCH1 than in DYT/PARK-TH [
80].