 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patrick C. Bradshaw | -- | 6878 | 2022-08-30 18:53:18 | | | |
| 2 | Lindsay Dong | -38 word(s) | 6840 | 2022-08-31 03:35:34 | | |
Video Upload Options
Dysfunctional mitochondrial quality control (MQC) is implicated in the pathogenesis of Parkinson’s disease (PD). The improper selection of mitochondria for mitophagy increases reactive oxygen species (ROS) levels and lowers ATP levels. The downstream effects include oxidative damage, failure to maintain proteostasis and ion gradients, and decreased NAD+ and NADPH levels, resulting in insufficient energy metabolism and neurotransmitter synthesis. A ketosis-based metabolic therapy that increases the levels of (R)-3-hydroxybutyrate (BHB) may reverse the dysfunctional MQC by partially replacing glucose as an energy source, by stimulating mitophagy, and by decreasing inflammation. Fasting can potentially raise cytoplasmic NADPH levels by increasing the mitochondrial export and cytoplasmic metabolism of ketone body-derived citrate that increases flux through isocitrate dehydrogenase 1 (IDH1). NADPH is an essential cofactor for nitric oxide synthase, and the nitric oxide synthesized can diffuse into the mitochondrial matrix and react with electron transport chain-synthesized superoxide to form peroxynitrite. Excessive superoxide and peroxynitrite production can cause the opening of the mitochondrial permeability transition pore (mPTP) to depolarize the mitochondria and activate PINK1-dependent mitophagy. Both fasting and exercise increase ketogenesis and increase the cellular NAD+/NADH ratio, both of which are beneficial for neuronal metabolism.
1. Targeting MQC in PD
2. Interventional Strategies to Correct Dysfunctional MQC and Downstream Processes
2.1. NADPH Is Required for Proper MQC
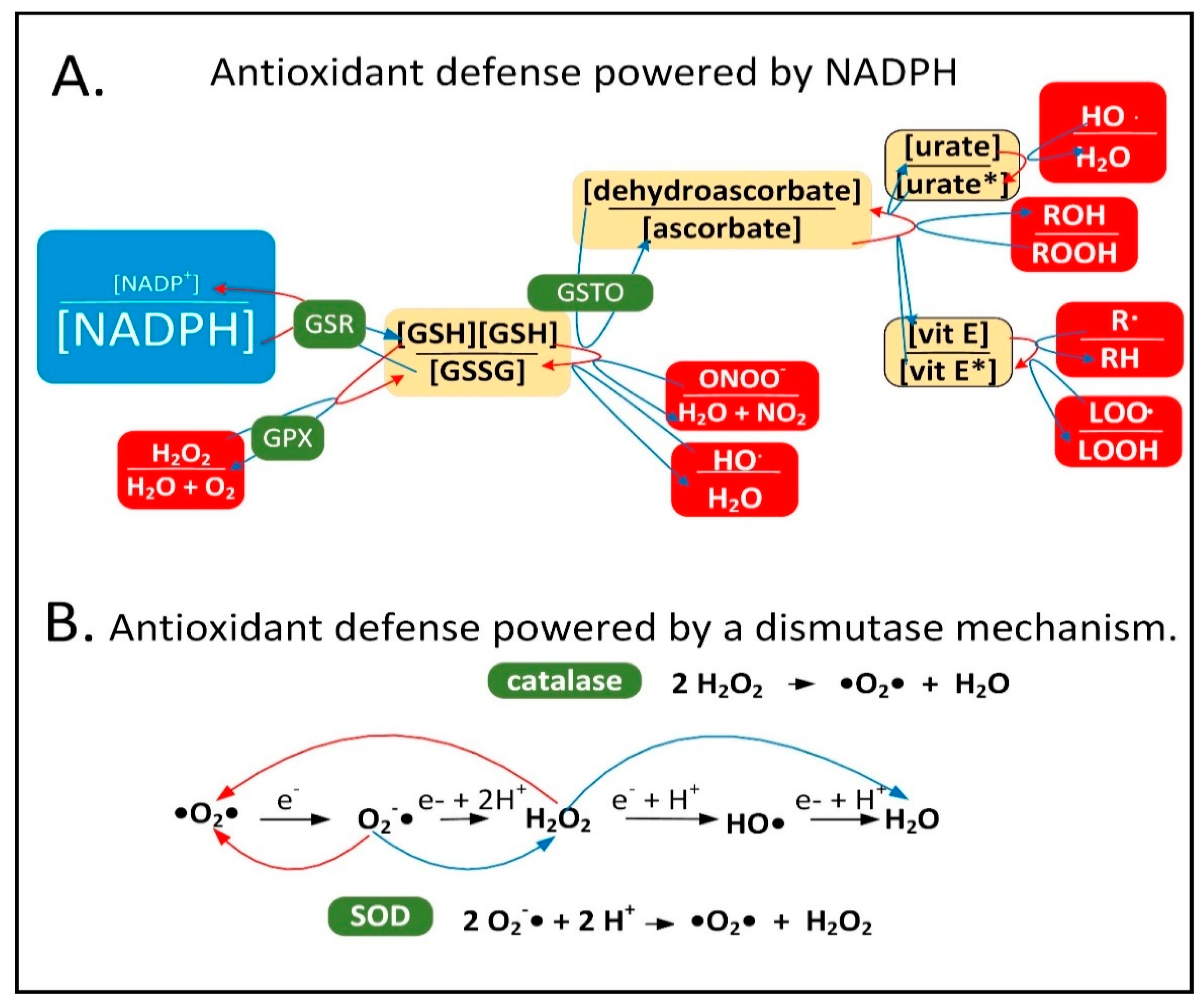
2.2. Mechanisms of Mitochondrial Protein Turnover
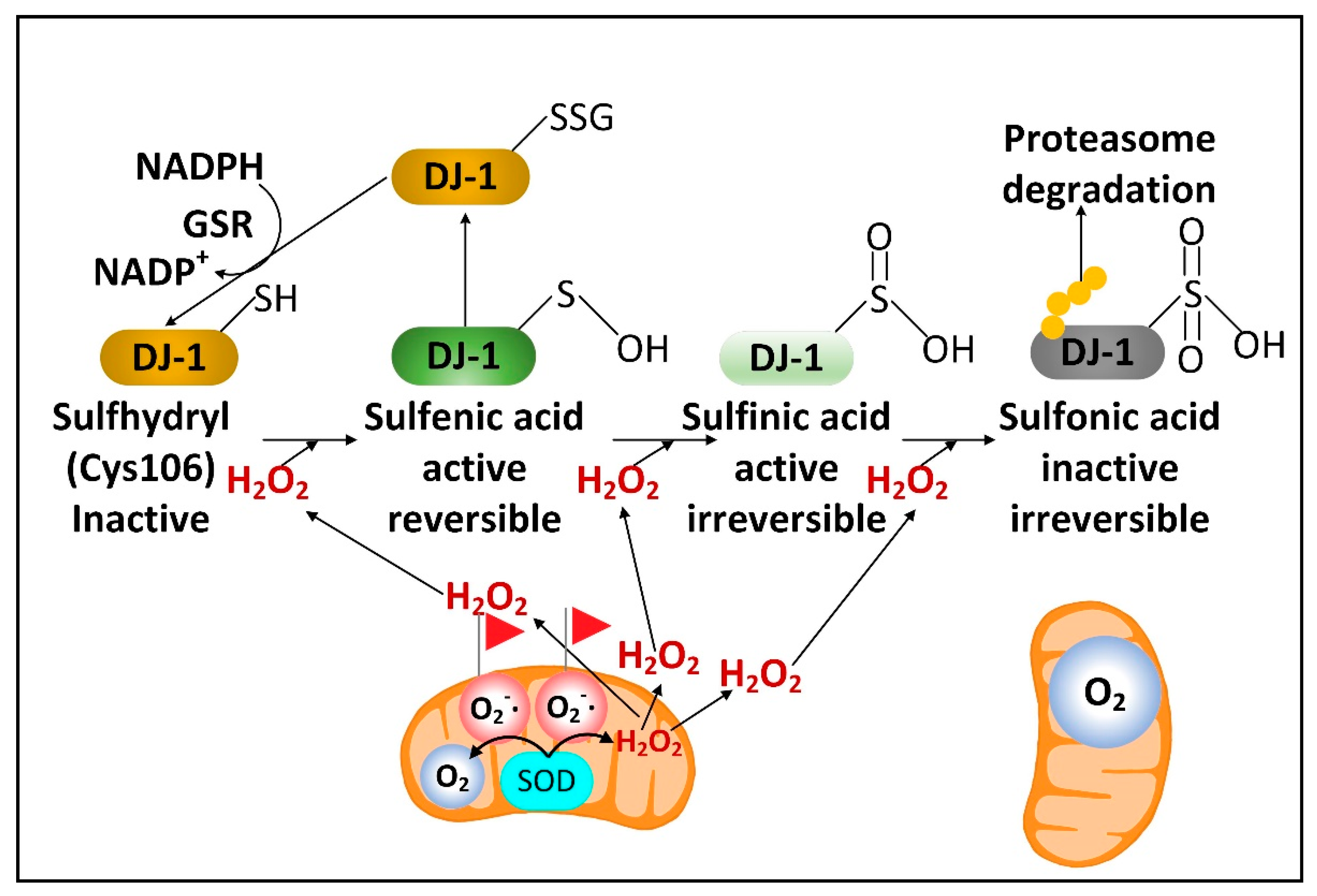
2.3. DJ-1 Is Regulated by Cytoplasmic NADPH and Glutathione and Controls Nrf2 Activation in Astrocytes
2.4. Fasting Increases the Hepatic Levels of ATF4 to Increase NADPH, FGF21, and Parkin
2.5. Fasting Activates Glutamate Dehydrogenase to Increase the Levels of the Anti-Aging Metabolite αkg
2.6. Fasting Decreases the Cytoplasmic [NADP+]/[NADPH] Ratio in Liver, but Its Effects on Neural Cell [NADP+]/[NADPH] Ratios Are Not Yet Known
2.7. Fasting May Improve PD Symptoms by Decreasing the Number of Senescent Astrocytes and Microglia without Affecting Substantia Nigra Mitochondrial DNA Deletion Levels
2.8. A Requirement for Fasting in Dietary Restriction-Induced Longevity
2.9. Evidence That Combining Fasting and Exercise Is Neuroprotective
2.10. Exercise and Its Effects on PD Brain and Muscles
2.11. Greater Metabolic Changes Induced by Exercise When the Exercise Occurs Early in the Active Period of the 24 h Circadian Cycle
Several protective compounds or “exerkines” were increased more during exercise early in the active phase, including serum AMP and beta-aminoisobutyric acid (BAIBA); muscle αkg, kynurenine, and GABA; liver BAIBA, kynurenine, and kynurenate; and hypothalamic BAIBA [83]. There was also increased muscle GSSG/GSH that occurred when exercise was carried out in the active phase. The more oxidized cellular environment following exercise in the active phase likely allows for more mitohormesis and ROS-induced Nrf2 activation several hours later, increasing NADPH levels and correcting the deficit. The most striking finding was the much greater global increase in 2-hydroxybutyrate (α-hydroxybutyrate) levels following active-phase exercise. In the same way that the pyruvate/lactate ratio is a marker of the cytoplasmic [NAD+]/[NADH] ratio due to the high activity of lactate dehydrogenase in most tissues [84], the 2-ketobutyrate/2-hydroxybutyrate ratio is also a global redox marker of the cytoplasmic [NAD+]/[NADH] ratio [85] due to 2-ketobutyrate and 2-hydroxybutyrate also serving as substrates for lactate dehydrogenase [86]. The more reduced [NAD+]/[NADH] ratio directly following exercise early in the active phase is likely an important signal to induce cytoprotective gene expression and metabolic changes leading to the restoration of the normal redox state.
2.12. Entrainment of the Circadian-Regulated Production of NAD+ in the Morning
2.13. Circadian Regulation of Mitophagy and Mitochondrial Dynamics May Play a Role in PD
3. Dopaminergic Neurons Are Vulnerable to Dysfunctional MQC
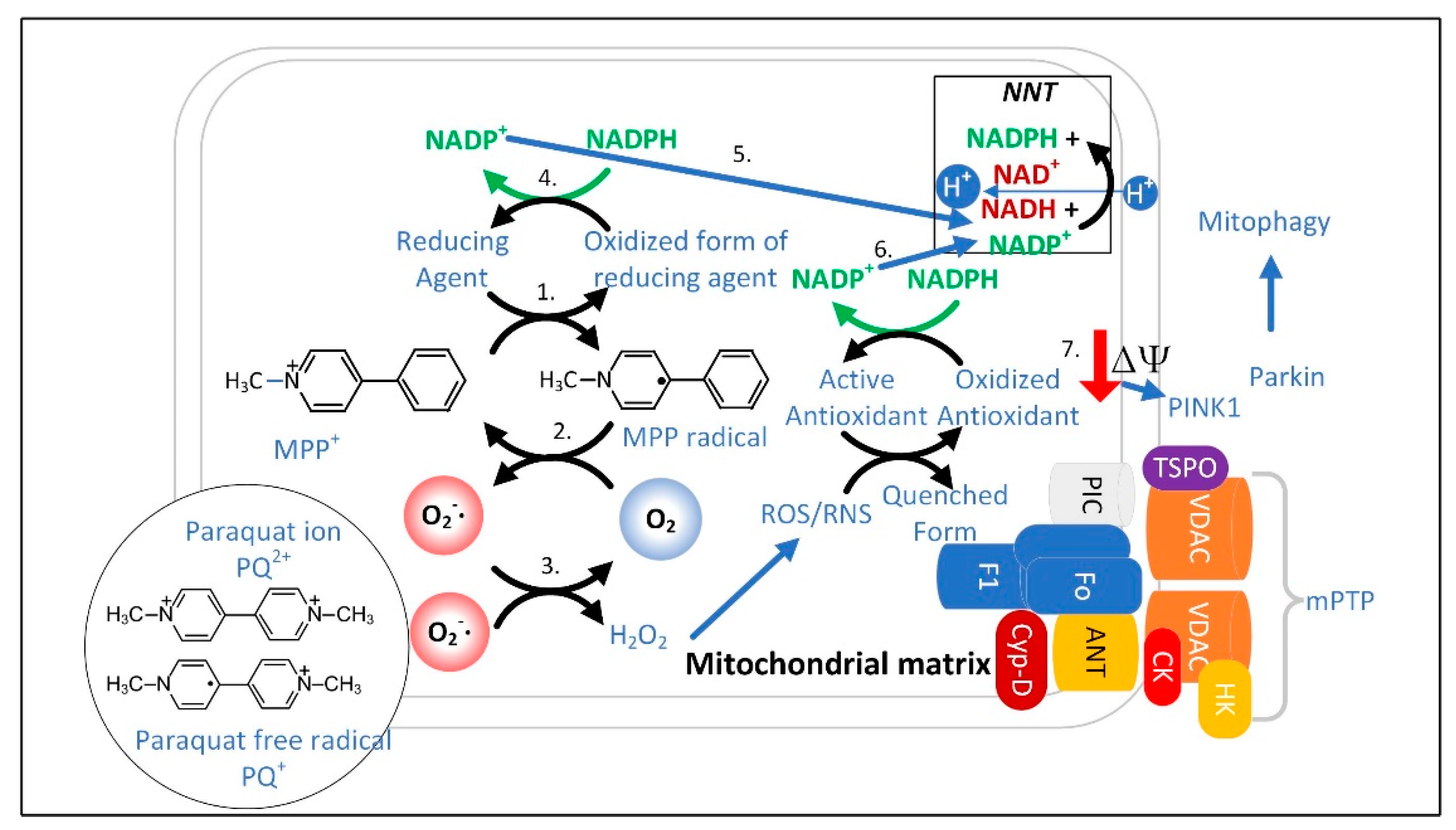
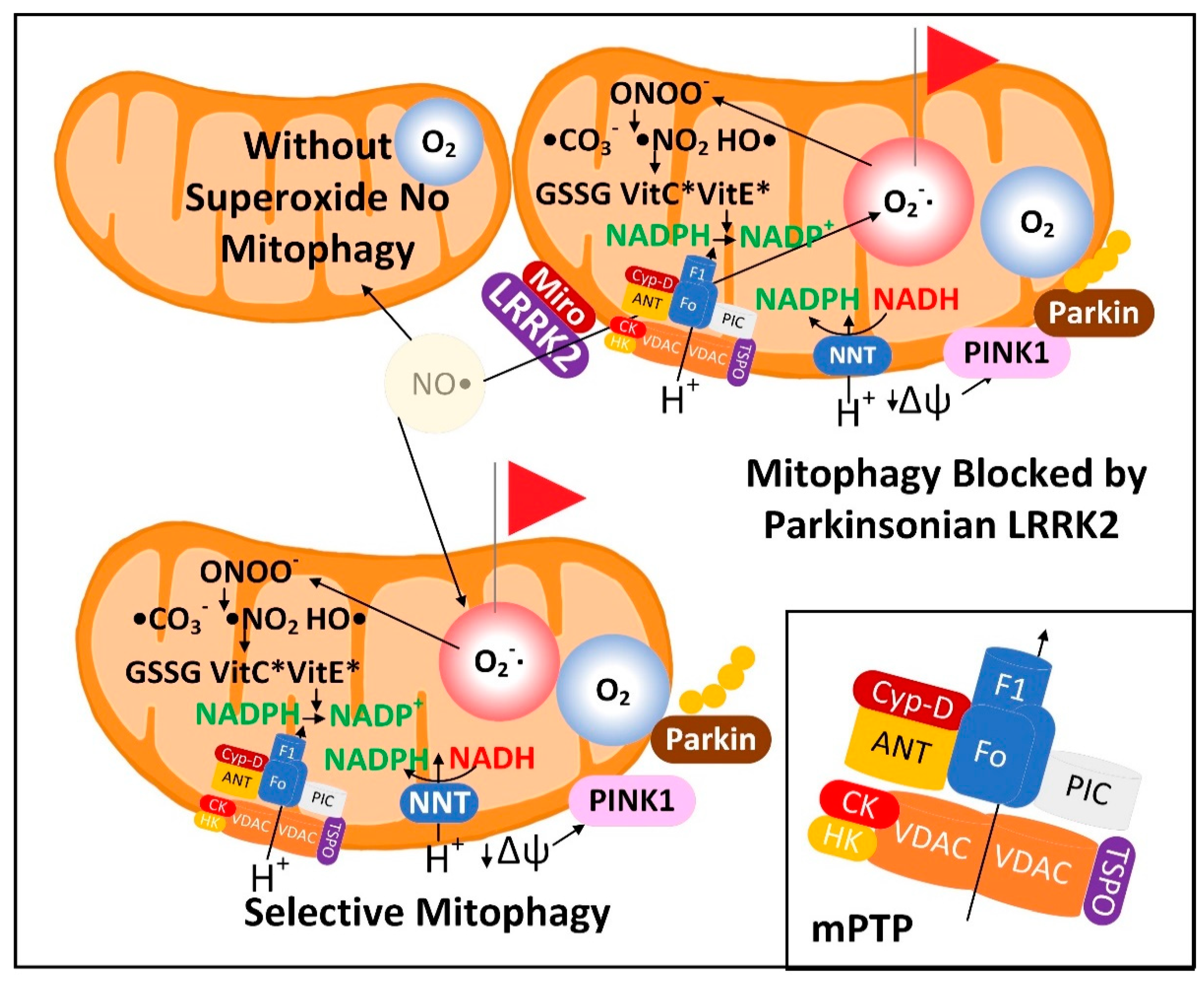
3.1. The Superoxide Sentinel Hypothesis of MQC
3.2. Neurons with Low-Quality Damaged Mitochondria Likely Show Decreased Proteolysis of α-Synuclein
3.3. Limited Clearance of ROS/RNS Leads to the Oxidation and Depletion of Cardiolipin and Plasmalogens
3.4. NADPH May Decrease in PD Neurons and Glia Limiting the Synthesis of Serotonin, Melatonin, Epinephrine, Norepinephrine, and •NO
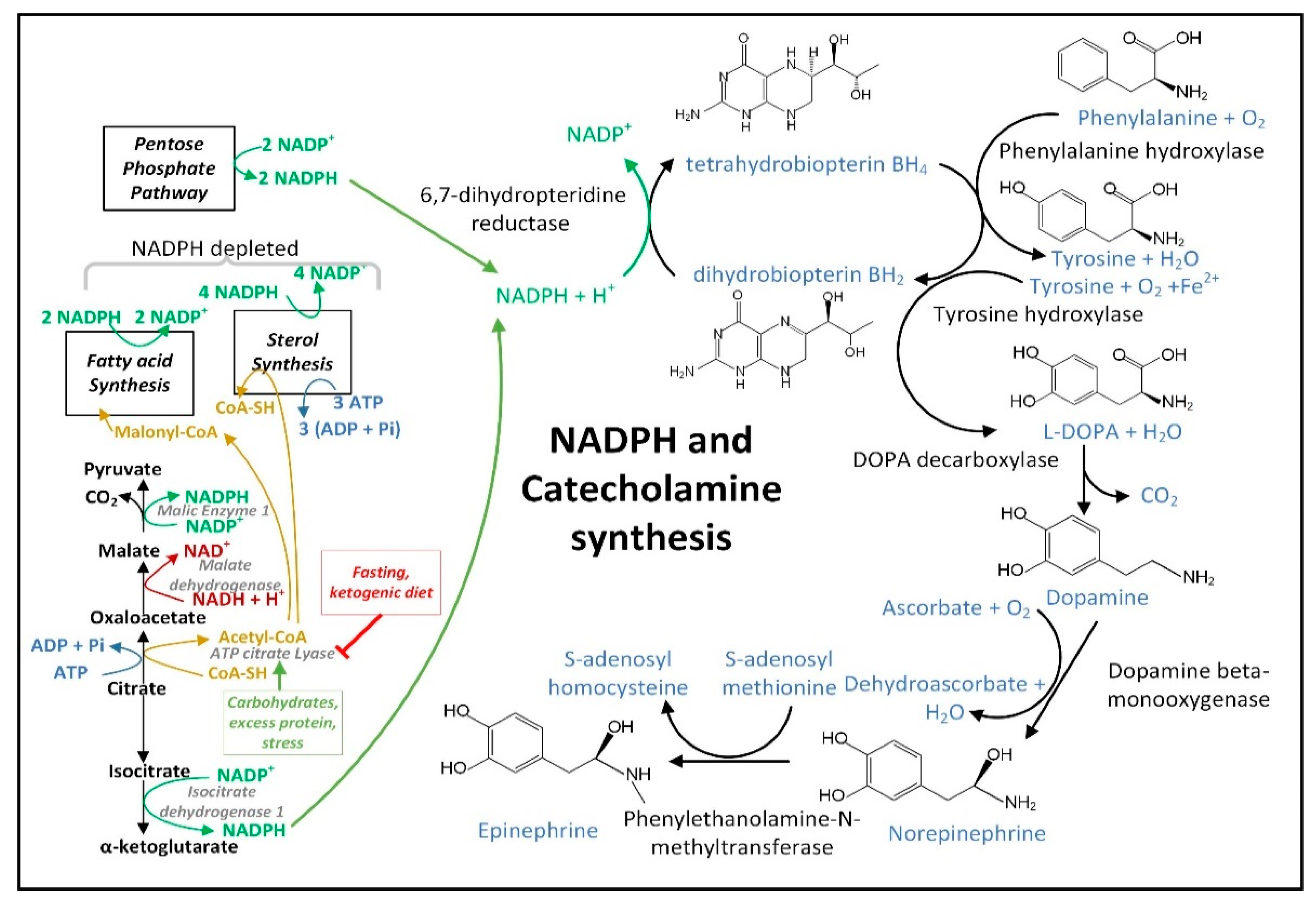
3.5. Insulin Released after Feeding Increases Cytoplasmic NADPH Oxidation by Stimulating Fatty Acid Synthesis
The decreased hepatic cytoplasmic [NADP+]/[NADPH] ratio that occurs during fasting may require glucagon [140] and may not occur after carbohydrate consumption in part due to insulin signaling. When insulin binds its receptor on the plasma membrane, a signaling cascade is initiated activating protein kinase AKT that phosphorylates ATP-citrate lyase (ACLY) serine 454 to activate the enzyme [141]. Thus, the citrate that is exported from the mitochondrial matrix is diverted away from the citrate–αkg shuttle that uses IDH1 for NADPH synthesis to boost cytoplasmic NADPH levels, and instead it is routed to the citrate–malate or citrate–pyruvate shuttles that use ACLY for the synthesis of acetyl-CoA and oxaloacetate. Acetyl-CoA is largely used for NADPH-depleting fatty acid synthesis. The malate dehydrogenase 1 (MDH1) enzyme converts the generated oxaloacetate into malate, which can either be transported back into the mitochondrial matrix for the citrate–malate shuttle or converted to pyruvate by cytoplasmic ME1 with the concurrent reduction of NADP+ to NADPH as part of the citrate–pyruvate shuttle. The pyruvate is then transported back into the mitochondrial matrix.
3.6. Experiments Are Needed to Determine the (Free) Cytoplasmic [NADP+]/[NADPH] Ratio in Neural Cells from Aged and PD Model Mice and How These Are Altered by Fasting
3.7. Dysfunctional MQC Leads to Loss of Circadian Entrainment of NAD+ Synthesis
4. Conclusions
References
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The Cell Biology of Parkinson’s Disease. J. Cell Biol. 2021, 220, e202012095.
- Singh, F.; Ganley, I.G. Parkinson’s Disease and Mitophagy: An Emerging Role for LRRK2. Biochem. Soc. Trans. 2021, 49, 551–562.
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The Pharmacological Regulation of Cellular Mitophagy. Nat. Chem. Biol. 2017, 13, 136–146.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211.
- Butkovich, L.M.; Houser, M.C.; Chalermpalanupap, T.; Porter-Stransky, K.A.; Iannitelli, A.F.; Boles, J.S.; Lloyd, G.M.; Coomes, A.S.; Eidson, L.N.; De Sousa Rodrigues, M.E.; et al. Transgenic Mice Expressing Human α-Synuclein in Noradrenergic Neurons Develop Locus Ceruleus Pathology and Nonmotor Features of Parkinson’s Disease. J. Neurosci. 2020, 40, 7559–7576.
- McCarty, M.F.; Lerner, A. Nutraceuticals Targeting Generation and Oxidant Activity of Peroxynitrite May Aid Prevention and Control of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3624.
- Rodriguez-Enriquez, S.; He, L.; Lemasters, J.J. Role of Mitochondrial Permeability Transition Pores in Mitochondrial Autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2463–2472.
- Lemasters, J.J.; Nieminen, A.-L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The Mitochondrial Permeability Transition in Cell Death: A Common Mechanism in Necrosis, Apoptosis and Autophagy. Biochim. Biophys. Acta Bioenerg. 1998, 1366, 177–196.
- Salvi, M.; Battaglia, V.; Brunati, A.M.; Rocca, N.L.; Tibaldi, E.; Pietrangeli, P.; Marcocci, L.; Mondovi, B.; Rossi, C.A.; Toninello, A. Catalase Takes Part in Rat Liver Mitochondria Oxidative Stress Defense. J. Biol. Chem. 2007, 282, 24407–24415.
- Radi, R.; Turrens, J.F.; Chang, L.Y.; Bush, K.M.; Crapo, J.D.; Freeman, B.A. Detection of Catalase in Rat Heart Mitochondria. J. Biol. Chem. 1991, 266, 22028–22034.
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666.
- Lamberts, J.T.; Hildebrandt, E.N.; Brundin, P. Spreading of α-Synuclein in the Face of Axonal Transport Deficits in Parkinson’s Disease: A Speculative Synthesis. Neurobiol. Dis. 2015, 77, 276–283.
- Gomez-Fabra Gala, M.; Vögtle, F. Mitochondrial Proteases in Human Diseases. FEBS Lett. 2021, 595, 1205–1222.
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide Is the Major Reactive Oxygen Species Regulating Autophagy. Cell Death Differ. 2009, 16, 1040–1052.
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The Expression of DJ-1 (PARK7) in Normal Human CNS and Idiopathic Parkinson’s Disease. Brain 2004, 127, 420–430.
- Blackinton, J.; Lakshminarasimhan, M.; Thomas, K.J.; Ahmad, R.; Greggio, E.; Raza, A.S.; Cookson, M.R.; Wilson, M.A. Formation of a Stabilized Cysteine Sulfinic Acid Is Critical for the Mitochondrial Function of the Parkinsonism Protein DJ-1. J. Biol. Chem. 2009, 284, 6476–6485.
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The Multifaceted Role of Nrf2 in Mitochondrial Function. Curr. Opin. Toxicol. 2016, 1, 80–91.
- Yao, W.; Lin, S.; Su, J.; Cao, Q.; Chen, Y.; Chen, J.; Zhang, Z.; Hashimoto, K.; Qi, Q.; Zhang, J. Activation of BDNF by Transcription Factor Nrf2 Contributes to Antidepressant-like Actions in Rodents. Transl. Psychiatry 2021, 11, 140.
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the Mechanism of Pathogenesis of Parkinson’s Disease. J. Bioenerg. Biomembr. 2019, 51, 175–188.
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant Stress Evoked by Pacemaking in Dopaminergic Neurons Is Attenuated by DJ-1. Nature 2010, 468, 696–700.
- Xu, S.; Yang, X.; Qian, Y.; Xiao, Q. Parkinson’s Disease-Related DJ-1 Modulates the Expression of Uncoupling Protein 4 against Oxidative Stress. J. Neurochem. 2018, 145, 312–322.
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.K.; Alam, J. Identification of Activating Transcription Factor 4 (ATF4) as an Nrf2-Interacting Protein: Implication for Heme Oxygenase-1 Gene Regulation. J. Biol. Chem. 2001, 276, 20858–20865.
- Zong, Z.-H.; Du, Z.-X.; Li, N.; Li, C.; Zhang, Q.; Liu, B.-Q.; Guan, Y.; Wang, H.-Q. Implication of Nrf2 and ATF4 in Differential Induction of CHOP by Proteasome Inhibition in Thyroid Cancer Cells. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1395–1404.
- Almeida, L.M.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. The PERKs of Mitochondria Protection during Stress: Insights for PERK Modulation in Neurodegenerative and Metabolic Diseases. Biol. Rev. Camb. Philos. Soc. 2022; early view.
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.-W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 Is an Oxidative Stress–Inducible, Prodeath Transcription Factor in Neurons in Vitro and in Vivo. J. Exp. Med. 2008, 205, 1227–1242.
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-Omics Analysis Identifies ATF4 as a Key Regulator of the Mitochondrial Stress Response in Mammals. J. Cell Biol. 2017, 216, 2027–2045.
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 Pathway and Its Cross Talk with Nrf2 in Mitochondrial Quality Control. J. Clin. Biochem. Nutr. 2019, 64, 18–37.
- Torrence, M.E.; MacArthur, M.R.; Hosios, A.M.; Valvezan, A.J.; Asara, J.M.; Mitchell, J.R.; Manning, B.D. The MTORC1-Mediated Activation of ATF4 Promotes Protein and Glutathione Synthesis Downstream of Growth Signals. eLife 2021, 10, e63326.
- Renz, P.F.; Valdivia-Francia, F.; Sendoel, A. Some like It Translated: Small ORFs in the 5′UTR. Exp. Cell Res. 2020, 396, 112229.
- Endo, J.; Sano, M.; Katayama, T.; Hishiki, T.; Shinmura, K.; Morizane, S.; Matsuhashi, T.; Katsumata, Y.; Zhang, Y.; Ito, H.; et al. Metabolic Remodeling Induced by Mitochondrial Aldehyde Stress Stimulates Tolerance to Oxidative Stress in the Heart. Circ. Res. 2009, 105, 1118–1127.
- Wang, X.; Zhang, G.; Dasgupta, S.; Niewold, E.L.; Li, C.; Li, Q.; Luo, X.; Tan, L.; Ferdous, A.; Lorenzi, P.L.; et al. ATF4 Protects the Heart From Failure by Antagonizing Oxidative Stress. Circ. Res. 2022, 131, 91–105.
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. MTORC1 Induces Purine Synthesis Through Control of the Mitochondrial Tetrahydrofolate Cycle. Science 2016, 351, 728–733.
- Hill, C.M.; Albarado, D.C.; Coco, L.G.; Spann, R.A.; Khan, M.S.; Qualls-Creekmore, E.; Burk, D.H.; Burke, S.J.; Collier, J.J.; Yu, S.; et al. FGF21 Is Required for Protein Restriction to Extend Lifespan and Improve Metabolic Health in Male Mice. Nat. Commun. 2022, 13, 1897.
- Zhang, Y.; Xie, Y.; Berglund, E.D.; Coate, K.C.; He, T.T.; Katafuchi, T.; Xiao, G.; Potthoff, M.J.; Wei, W.; Wan, Y.; et al. The Starvation Hormone, Fibroblast Growth Factor-21, Extends Lifespan in Mice. eLife 2012, 1, e00065.
- Klaus, S.; Igual Gil, C.; Ost, M. Regulation of Diurnal Energy Balance by Mitokines. Cell Mol. Life Sci. 2021, 78, 3369–3384.
- Kim, K.H.; Kim, S.H.; Min, Y.-K.; Yang, H.-M.; Lee, J.-B.; Lee, M.-S. Acute Exercise Induces FGF21 Expression in Mice and in Healthy Humans. PLoS ONE 2013, 8, e63517.
- Wan, X.; Lu, X.; Xiao, Y.; Lin, Y.; Zhu, H.; Ding, T.; Yang, Y.; Huang, Y.; Zhang, Y.; Liu, Y.-L.; et al. ATF4- and CHOP-Dependent Induction of FGF21 through Endoplasmic Reticulum Stress. BioMed Res. Int. 2014, 2014, e807874.
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin Is Transcriptionally Regulated by ATF4: Evidence for an Interconnection between Mitochondrial Stress and ER Stress. Cell Death. Differ. 2011, 18, 769–782.
- Li, W.; Li, X.; Miller, R.A. ATF4 Activity: A Common Feature Shared by Many Kinds of Slow-Aging Mice. Aging Cell 2014, 13, 1012–1018.
- Koyanagi, S.; Hamdan, A.M.; Horiguchi, M.; Kusunose, N.; Okamoto, A.; Matsunaga, N.; Ohdo, S. CAMP-Response Element (CRE)-Mediated Transcription by Activating Transcription Factor-4 (ATF4) Is Essential for Circadian Expression of the Period2 Gene. J. Biol. Chem. 2011, 286, 32416–32423.
- Pathak, S.S.; Liu, D.; Li, T.; de Zavalia, N.; Zhu, L.; Li, J.; Karthikeyan, R.; Alain, T.; Liu, A.C.; Storch, K.-F.; et al. The EIF2α Kinase GCN2 Modulates Period and Rhythmicity of the Circadian Clock by Translational Control of Atf4. Neuron 2019, 104, 724–735.e6.
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic β Cells. Cell 2006, 126, 941–954.
- Shaw, E.; Talwadekar, M.; Rashida, Z.; Mohan, N.; Acharya, A.; Khatri, S.; Laxman, S.; Kolthur-Seetharam, U. Anabolic SIRT4 Exerts Retrograde Control over TORC1 Signaling by Glutamine Sparing in the Mitochondria. Mol. Cell Biol. 2020, 40, e00212-19.
- de Goede, P.; Wüst, R.C.I.; Schomakers, B.V.; Denis, S.; Vaz, F.M.; Pras-Raves, M.L.; van Weeghel, M.; Yi, C.-X.; Kalsbeek, A.; Houtkooper, R.H. Time-Restricted Feeding during the Inactive Phase Abolishes the Daily Rhythm in Mitochondrial Respiration in Rat Skeletal Muscle. FASEB J. 2022, 36, e22133.
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The Metabolite Alpha-Ketoglutarate Extends Lifespan by Inhibiting the ATP Synthase and TOR. Nature 2014, 510, 397–401.
- Su, Y.; Wang, T.; Wu, N.; Li, D.; Fan, X.; Xu, Z.; Mishra, S.K.; Yang, M. Alpha-Ketoglutarate Extends Drosophila Lifespan by Inhibiting MTOR and Activating AMPK. Aging 2019, 11, 4183–4197.
- Shahmirzadi, A.A.; Edgar, D.; Liao, C.-Y.; Hsu, Y.-M.; Lucanic, M.; Shahmirzadi, A.A.; Wiley, C.D.; Gan, G.; Kim, D.E.; Kasler, H.G.; et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020, 32, 447–456.e6.
- Harrison, A.P.; Pierzynowski, S.G. Biological Effects of 2-Oxoglutarate with Particular Emphasis on the Regulation of Protein, Mineral and Lipid Absorption/Metabolism, Muscle Performance, Kidney Function, Bone Formation and Cancerogenesis, All Viewed from a Healthy Ageing Perspective State of the Art—Review Article. J. Physiol. Pharmacol. 2008, 59, 91–106.
- Lin, A.-L.; Zhang, W.; Gao, X.; Watts, L. Caloric Restriction Increases Ketone Bodies Metabolism and Preserves Blood Flow in Aging Brain. Neurobiol. Aging 2015, 36, 2296–2303.
- Moebus, S.; Göres, L.; Lösch, C.; Jöckel, K.-H. Impact of Time since Last Caloric Intake on Blood Glucose Levels. Eur. J. Epidemiol. 2011, 26, 719–728.
- Gannon, M.C.; Nuttall, F.Q.; Lane, J.T.; Fang, S.; Gupta, V.; Sandhofer, C.R. Effect of 24 Hours of Starvation on Plasma Glucose and Insulin Concentrations in Subjects with Untreated Non—Insulin-Dependent Diabetes Mellitus. Metabolism 1996, 45, 492–497.
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F. Liver and Kidney Metabolism during Prolonged Starvation. J. Clin. Investig. 1969, 48, 574–583.
- Cahill, G.F., Jr. Fuel Metabolism in Starvation. Annu. Rev. Nutr. 2006, 26, 1–22.
- Minich, T.; Yokota, S.; Dringen, R. Cytosolic and Mitochondrial Isoforms of NADP+-Dependent Isocitrate Dehydrogenases Are Expressed in Cultured Rat Neurons, Astrocytes, Oligodendrocytes and Microglial Cells. J. Neurochem. 2003, 86, 605–614.
- Kurz, G.M.; Wiesinger, H.; Hamprecht, B. Purification of Cytosolic Malic Enzyme from Bovine Brain, Generation of Monoclonal Antibodies, and Immunocytochemical Localization of the Enzyme in Glial Cells of Neural Primary Cultures. J. Neurochem. 1993, 60, 1467–1474.
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Cerebrospinal and Blood Levels of Amino Acids as Potential Biomarkers for Parkinson’s Disease: Review and Meta-analysis. Eur. J. Neurol. 2020, 27, 2336–2347.
- Verma, D.K.; Seo, B.A.; Ghosh, A.; Ma, S.-X.; Hernandez-Quijada, K.; Andersen, J.K.; Ko, H.S.; Kim, Y.-H. Alpha-Synuclein Preformed Fibrils Induce Cellular Senescence in Parkinson’s Disease Models. Cells 2021, 10, 1694.
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940.
- Martini, H.; Passos, J.F. Cellular Senescence: All Roads Lead to Mitochondria. FEBS J. 2022; early view.
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced MtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917.
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA Fragments Exit Mitochondria via MPTP- and VDAC-Dependent Channels to Activate NLRP3 Inflammasome and Interferon Signaling. Immunity, 2022, in press.
- Salnikov, V.; Lukyánenko, Y.O.; Frederick, C.A.; Lederer, W.J.; Lukyánenko, V. Probing the Outer Mitochondrial Membrane in Cardiac Mitochondria with Nanoparticles. Biophys. J. 2007, 92, 1058–1071.
- Rauckhorst, A.J.; Pfeiffer, D.R.; Broekemeier, K.M. The IPLA2γ Is Identified as the Membrane Potential Sensitive Phospholipase in Liver Mitochondria. FEBS Lett. 2015, 589, 2367–2371.
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A. Mitochondrial Permeability Transition Triggers the Release of MtDNA Fragments. CMLS Cell. Mol. Life Sci. 2004, 61, 3100–3103.
- García, N.; García, J.J.; Correa, F.; Chávez, E. The Permeability Transition Pore as a Pathway for the Release of Mitochondrial DNA. Life Sci. 2005, 76, 2873–2880.
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ Supplementation Reduces Neuroinflammation and Cell Senescence in a Transgenic Mouse Model of Alzheimer’s Disease via CGAS–STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118.
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular Mechanisms of Dietary Restriction Promoting Health and Longevity. Nat. Rev. Mol. Cell Biol. 2022, 23, 56–73.
- Mitchell, S.J.; Bernier, M.; Mattison, J.A.; Aon, M.A.; Kaiser, T.A.; Anson, R.M.; Ikeno, Y.; Anderson, R.M.; Ingram, D.K.; de Cabo, R. Daily Fasting Improves Health and Survival in Male Mice Independent of Diet Composition and Calories. Cell Metab. 2019, 29, 221–228.e3.
- Shimokawa, I.; Komatsu, T.; Hayashi, N.; Kim, S.-E.; Kawata, T.; Park, S.; Hayashi, H.; Yamaza, H.; Chiba, T.; Mori, R. The Life-Extending Effect of Dietary Restriction Requires Foxo3 in Mice. Aging Cell 2015, 14, 707–709.
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-Beta-Hydroxybutyrate Extends Lifespan in C. elegans. Aging 2014, 6, 621–644.
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent Metabolic Switching, Neuroplasticity and Brain Health. Nat. Rev. Neurosci. 2018, 19, 81–94.
- Hvingelby, V.S.; Glud, A.N.; Sørensen, J.C.H.; Tai, Y.; Andersen, A.S.M.; Johnsen, E.; Moro, E.; Pavese, N. Interventions to Improve Gait in Parkinson’s Disease: A Systematic Review of Randomized Controlled Trials and Network Meta-Analysis. J. Neurol. 2022, 269, 4068–4079.
- Uhrbrand, A.; Stenager, E.; Pedersen, M.S.; Dalgas, U. Parkinson’s Disease and Intensive Exercise Therapy—A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Neurol. Sci. 2015, 353, 9–19.
- Stillman, C.M.; Cohen, J.; Lehman, M.E.; Erickson, K.I. Mediators of Physical Activity on Neurocognitive Function: A Review at Multiple Levels of Analysis. Front. Hum. Neurosci. 2016, 10, 626.
- Di Benedetto, S.; Müller, L.; Wenger, E.; Düzel, S.; Pawelec, G. Contribution of Neuroinflammation and Immunity to Brain Aging and the Mitigating Effects of Physical and Cognitive Interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128.
- Simeone, T.A.; Simeone, K.A.; Rho, J.M. Ketone Bodies as Anti-Seizure Agents. Neurochem. Res. 2017, 42, 2011–2018.
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 Year Multidomain Intervention of Diet, Exercise, Cognitive Training, and Vascular Risk Monitoring versus Control to Prevent Cognitive Decline in at-Risk Elderly People (FINGER): A Randomised Controlled Trial. Lancet 2015, 385, 2255–2263.
- Kwon, J.H.; Moon, K.M.; Min, K.-W. Exercise-Induced Myokines Can Explain the Importance of Physical Activity in the Elderly: An Overview. Healthcare 2020, 8, 378.
- Hao, Z.; Zhang, X.; Chen, P. Effects of Ten Different Exercise Interventions on Motor Function in Parkinson’s Disease Patients—A Network Meta-Analysis of Randomized Controlled Trials. Brain Sci. 2022, 12, 698.
- Gibala, M.J.; Little, J.P.; MacDonald, M.J.; Hawley, J.A. Physiological Adaptations to Low-Volume, High-Intensity Interval Training in Health and Disease. J. Physiol. 2012, 590, 1077–1084.
- Wahl, P.; Bloch, W.; Proschinger, S. The Molecular Signature of High-Intensity Training in the Human Body. Int. J. Sports Med. 2022, 43, 195–205.
- Ma, J.K.; Scribbans, T.D.; Edgett, B.A.; Boyd, J.C.; Simpson, C.A.; Little, J.P.; Gurd, B.J. Extremely Low-Volume, High-Intensity Interval Training Improves Exercise Capacity and Increases Mitochondrial Protein Content in Human Skeletal Muscle. Open J. Mol. Integr. Physiol. 2013, 3, 202–210.
- Sato, S.; Dyar, K.A.; Treebak, J.T.; Jepsen, S.L.; Ehrlich, A.M.; Ashcroft, S.P.; Trost, K.; Kunzke, T.; Prade, V.M.; Small, L.; et al. Atlas of Exercise Metabolism Reveals Time-Dependent Signatures of Metabolic Homeostasis. Cell Metab. 2022, 34, 329–345.e8.
- Williamson, D.H.; Lund, P.; Krebs, H.A. The Redox State of Free Nicotinamide-Adenine Dinucleotide in the Cytoplasm and Mitochondria of Rat Liver. Biochem. J. 1967, 103, 514–527.
- Goodman, R.P.; Markhard, A.L.; Shah, H.; Sharma, R.; Skinner, O.S.; Clish, C.B.; Deik, A.; Patgiri, A.; Hsu, Y.-H.; Masia, R.; et al. Hepatic NADH Reductive Stress Underlies Common Variation in Metabolic Traits. Nature 2020, 583, 122–126.
- Naghizadeh, F. Oxidation of Alpha-Hydroxybutyrate by Human Serum. Clin. Chim. Acta 1977, 81, 277–282.
- Perrin, L.; Loizides-Mangold, U.; Chanon, S.; Gobet, C.; Hulo, N.; Isenegger, L.; Weger, B.D.; Migliavacca, E.; Charpagne, A.; Betts, J.A.; et al. Transcriptomic Analyses Reveal Rhythmic and CLOCK-Driven Pathways in Human Skeletal Muscle. eLife 2018, 7, e34114.
- Schug, T.T.; Li, X. Sirtuin 1 in Lipid Metabolism and Obesity. Ann. Med. 2011, 43, 198–211.
- Luna, A.; McFadden, G.B.; Aladjem, M.I.; Kohn, K.W. Predicted Role of NAD Utilization in the Control of Circadian Rhythms during DNA Damage Response. PLoS Comput. Biol. 2015, 11, e1004144.
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.-K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian Clock Feedback Cycle Through NAMPT-Mediated NAD+ Biosynthesis. Science 2009, 324, 651–654.
- van Moorsel, D.; Hansen, J.; Havekes, B.; Scheer, F.A.J.L.; Jörgensen, J.A.; Hoeks, J.; Schrauwen-Hinderling, V.B.; Duez, H.; Lefebvre, P.; Schaper, N.C.; et al. Demonstration of a Day-Night Rhythm in Human Skeletal Muscle Oxidative Capacity. Mol. Metab. 2016, 5, 635–645.
- Qiu, Z.; Ming, H.; Lei, S.; Zhou, B.; Zhao, B.; Yu, Y.; Xue, R.; Xia, Z. Roles of HDAC3-Orchestrated Circadian Clock Gene Oscillations in Diabetic Rats Following Myocardial Ischaemia/Reperfusion Injury. Cell Death Dis. 2021, 12, 43.
- Parekh, P.K.; Ozburn, A.R.; McClung, C.A. Circadian Clock Genes: Effects on Dopamine, Reward and Addiction. Alcohol 2015, 49, 341–349.
- Breen, D.P.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.M.; Reddy, A.B.; Barker, R.A. Sleep and Circadian Rhythm Regulation in Early Parkinson Disease. JAMA Neurol. 2014, 71, 589–595.
- Cai, Y.; Liu, S.; Sothern, R.B.; Xu, S.; Chan, P. Expression of Clock Genes Per1 and Bmal1 in Total Leukocytes in Health and Parkinson’s Disease: Clock Genes in PD. Eur. J. Neurol. 2010, 17, 550–554.
- Li, S.-Y.; Wang, Y.-L.; Liu, W.-W.; Lyu, D.-J.; Wang, F.; Mao, C.-J.; Yang, Y.-P.; Hu, L.-F.; Liu, C.-F. Long-Term Levodopa Treatment Accelerates the Circadian Rhythm Dysfunction in a 6-Hydroxydopamine Rat Model of Parkinson’s Disease. Chin. Med. J. 2017, 130, 1085–1092.
- Li, H.; Song, S.; Wang, Y.; Huang, C.; Zhang, F.; Liu, J.; Hong, J.-S. Low-Grade Inflammation Aggravates Rotenone Neurotoxicity and Disrupts Circadian Clock Gene Expression in Rats. Neurotox. Res. 2019, 35, 421–431.
- Liu, W.; Wei, S.; Huang, G.; Liu, L.; Gu, C.; Shen, Y.; Wang, X.; Xia, S.; Xie, A.; Hu, L.; et al. BMAL1 Regulation of Microglia-mediated Neuroinflammation in MPTP-induced Parkinson’s Disease Mouse Model. FASEB J. 2020, 34, 6570–6581.
- Kim, J.; Jang, S.; Choi, M.; Chung, S.; Choe, Y.; Choe, H.K.; Son, G.H.; Rhee, K.; Kim, K. Abrogation of the Circadian Nuclear Receptor REV-ERBα Exacerbates 6-Hydroxydopamine-Induced Dopaminergic Neurodegeneration. Mol. Cells 2018, 41, 742–752.
- de Goede, P.; Wefers, J.; Brombacher, E.C.; Schrauwen, P.; Kalsbeek, A. Circadian Rhythms in Mitochondrial Respiration. J. Mol. Endocrinol. 2018, 60, R115–R130.
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720.
- Schmitt, K.; Grimm, A.; Dallmann, R.; Oettinghaus, B.; Restelli, L.M.; Witzig, M.; Ishihara, N.; Mihara, K.; Ripperger, J.A.; Albrecht, U.; et al. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 2018, 27, 657–666.e5.
- Rabinovich-Nikitin, I.; Rasouli, M.; Reitz, C.J.; Posen, I.; Margulets, V.; Dhingra, R.; Khatua, T.N.; Thliveris, J.A.; Martino, T.A.; Kirshenbaum, L.A. Mitochondrial Autophagy and Cell Survival Is Regulated by the Circadian Clock Gene in Cardiac Myocytes during Ischemic Stress. Autophagy 2021, 17, 3794–3812.
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of Class I Histone Deacetylases Unveils a Mitochondrial Signature and Enhances Oxidative Metabolism in Skeletal Muscle and Adipose Tissue. Diabetes 2013, 62, 732–742.
- Shen, R.-S.; Abell, C.W.; Gessner, W.; Brossi, A. Serotonergic Conversion of MPTP and Dopaminergic Accumulation of MPP+. FEBS Lett. 1985, 189, 225–230.
- Ramsay, R.R.; Dadgar, J.; Trevor, A.; Singer, T.P. Energy-Driven Uptake of N-Methyl-4-Phenylpyridine by Brain Mitochondria Mediates the Neurotoxicity of MPTP. Life Sci. 1986, 39, 581–588.
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100.
- Cassarino, D.S.; Parks, J.K.; Parker, W.D.; Bennett, J.P. The Parkinsonian Neurotoxin MPP+ Opens the Mitochondrial Permeability Transition Pore and Releases Cytochrome c in Isolated Mitochondria via an Oxidative Mechanism. Biochim. Biophys. Acta Mol. Basis Dis. 1999, 1453, 49–62.
- Klaidman, L.K.; Adams, J.D.; Leung, A.C.; Sam Kim, S.; Cadenas, E. Redox Cycling of MPP+: Evidence for a New Mechanism Involving Hydride Transfer with Xanthine Oxidase, Aldehyde Dehydrogenase, and Lipoamide Dehydrogenase. Free. Radic. Biol. Med. 1993, 15, 169–179.
- Adams, J.D., Jr.; Klaidman, L.K.; Leung, A.C. MPP+ and MPDP+ Induced Oxygen Radical Formation with Mitochondrial Enzymes. Free Radic. Biol. Med. 1993, 15, 181–186.
- Veech, R.L.; Todd King, M.; Pawlosky, R.; Kashiwaya, Y.; Bradshaw, P.C.; Curtis, W. The “Great” Controlling Nucleotide Coenzymes. IUBMB Life 2019, 71, 565–579.
- Makino, K.; Hagiwara, T.; Murakami, A. A Mini Review: Fundamental Aspects of Spin Trapping with DMPO. Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1991, 37, 657–665.
- Hoehne, M.N.; Jacobs, L.J.H.C.; Lapacz, K.J.; Calabrese, G.; Murschall, L.M.; Marker, T.; Kaul, H.; Trifunovic, A.; Morgan, B.; Fricker, M.; et al. Spatial and Temporal Control of Mitochondrial H2O2 Release in Intact Human Cells. EMBO J. 2022, 41, e109169.
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.-L.; Tan, E.-K. Superoxide Drives Progression of Parkin/PINK1-Dependent Mitophagy Following Translocation of Parkin to Mitochondria. Cell Death Dis. 2017, 8, e3097.
- Zhang, J.; Wang, B.; Wang, H.; He, H.; Wu, Q.; Qin, X.; Yang, X.; Chen, L.; Xu, G.; Yuan, Z.; et al. Disruption of the Superoxide Anions-Mitophagy Regulation Axis Mediates Copper Oxide Nanoparticles-Induced Vascular Endothelial Cell Death. Free Radic. Biol. Med. 2018, 129, 268–278.
- Aon, M.A.; Cortassa, S.; Marbán, E.; O’Rourke, B. Synchronized Whole Cell Oscillations in Mitochondrial Metabolism Triggered by a Local Release of Reactive Oxygen Species in Cardiac Myocytes. J. Biol. Chem. 2003, 278, 44735–44744.
- Marchissio, M.J.; Francés, D.E.A.; Carnovale, C.E.; Marinelli, R.A. Mitochondrial Aquaporin-8 Knockdown in Human Hepatoma HepG2 Cells Causes ROS-Induced Mitochondrial Depolarization and Loss of Viability. Toxicol. Appl. Pharmacol. 2012, 264, 246–254.
- Almasalmeh, A.; Krenc, D.; Wu, B.; Beitz, E. Structural Determinants of the Hydrogen Peroxide Permeability of Aquaporins. FEBS J. 2014, 281, 647–656.
- Wang, Y.; Yen, F.S.; Zhu, X.G.; Timson, R.C.; Weber, R.; Xing, C.; Liu, Y.; Allwein, B.; Luo, H.; Yeh, H.-W.; et al. SLC25A39 Is Necessary for Mitochondrial Glutathione Import in Mammalian Cells. Nature 2021, 599, 136–140.
- Gialluisi, A.; Reccia, M.G.; Modugno, N.; Nutile, T.; Lombardi, A.; Di Giovannantonio, L.G.; Pietracupa, S.; Ruggiero, D.; Scala, S.; Gambardella, S.; et al. Identification of Sixteen Novel Candidate Genes for Late Onset Parkinson’s Disease. Mol. Neurodegener. 2021, 16, 35.
- Francisco, A.; Engel, D.F.; Figueira, T.R.; Rogério, F.; de Bem, A.F.; Castilho, R.F. Mitochondrial NAD(P)+ Transhydrogenase Is Unevenly Distributed in Different Brain Regions, and Its Loss Causes Depressive-like Behavior and Motor Dysfunction in Mice. Neuroscience 2020, 440, 210–229.
- Lopes da Fonseca, T.; Villar-Piqué, A.; Outeiro, T.F. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 2015, 5, 435–471.
- Peth, A.; Nathan, J.A.; Goldberg, A.L. The ATP Costs and Time Required to Degrade Ubiquitinated Proteins by the 26 S Proteasome. J. Biol. Chem. 2013, 288, 29215–29222.
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-Dependent Steps in the Binding of Ubiquitin Conjugates to the 26S Proteasome That Commit to Degradation. Mol. Cell 2010, 40, 671–681.
- Singh, R.; Cuervo, A.M. Autophagy in the Cellular Energetic Balance. Cell Metab. 2011, 13, 495–504.
- Benderdour, M.; Charron, G.; deBlois, D.; Comte, B.; Rosiers, C.D. Cardiac Mitochondrial NADP+-Isocitrate Dehydrogenase Is Inactivated through 4-Hydroxynonenal Adduct Formation: An Event that Precedes Hypertrophy Development. J. Biol. Chem. 2003, 278, 45154–45159.
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditis Elegans and Human Cancer Cells. Dev. Cell 2020, 54, 447–454.e4.
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis Driven by Radical Oxidation of N-6 Polyunsaturated Fatty Acids Mediates Acetaminophen-Induced Acute Liver Failure. Cell Death Dis. 2020, 11, 144.
- Tripathi, A.; Fanning, S.; Dettmer, U. Lipotoxicity Downstream of α-Synuclein Imbalance: A Relevant Pathomechanism in Synucleinopathies? Biomolecules 2021, 12, 40.
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1, 223–232.
- Jenkins, C.M.; Yang, K.; Liu, G.; Moon, S.H.; Dilthey, B.G.; Gross, R.W. Cytochrome c Is an Oxidative Stress–Activated Plasmalogenase That Cleaves Plasmenylcholine and Plasmenylethanolamine at the Sn-1 Vinyl Ether Linkage. J. Biol. Chem. 2018, 293, 8693–8709.
- Hsu, Y.-H.; Dumlao, D.S.; Cao, J.; Dennis, E.A. Assessing Phospholipase A2 Activity toward Cardiolipin by Mass Spectrometry. PLoS ONE 2013, 8, e59267.
- Mancuso, D.J.; Kotzbauer, P.; Wozniak, D.F.; Sims, H.F.; Jenkins, C.M.; Guan, S.; Han, X.; Yang, K.; Sun, G.; Malik, I.; et al. Genetic Ablation of Calcium-Independent Phospholipase A2γ Leads to Alterations in Hippocampal Cardiolipin Content and Molecular Species Distribution, Mitochondrial Degeneration, Autophagy, and Cognitive Dysfunction. J. Biol. Chem. 2009, 284, 35632–35644.
- Mahajan, M.; Bharambe, N.; Shang, Y.; Lu, B.; Mandal, A.; Madan Mohan, P.; Wang, R.; Boatz, J.C.; Manuel Martinez Galvez, J.; Shnyrova, A.V.; et al. NMR Identification of a Conserved Drp1 Cardiolipin-Binding Motif Essential for Stress-Induced Mitochondrial Fission. Proc. Natl. Acad. Sci. USA 2021, 118, e2023079118.
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination Signal for Mitophagy in Neuronal Cells. Nat. Cell Biol. 2013, 15, 1197–1205.
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323.
- Brekk, O.R.; Honey, J.R.; Lee, S.; Hallett, P.J.; Isacson, O. Cell Type-Specific Lipid Storage Changes in Parkinson’s Disease Patient Brains Are Recapitulated by Experimental Glycolipid Disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 27646–27654.
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.F.; Vercesi, A.E.; Castilho, R.F. A Spontaneous Mutation in the Nicotinamide Nucleotide Transhydrogenase Gene of C57BL/6J Mice Results in Mitochondrial Redox Abnormalities. Free. Radic. Biol. Med. 2013, 63, 446–456.
- Zhu, J.; Schwörer, S.; Berisa, M.; Kyung, Y.J.; Ryu, K.W.; Yi, J.; Jiang, X.; Cross, J.R.; Thompson, C.B. Mitochondrial NADP(H) Generation Is Essential for Proline Biosynthesis. Science 2021, 372, 968–972.
- Abraham, M.A.; Lam, T.K.T. Glucagon Action in the Brain. Diabetologia 2016, 59, 1367–1371.
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavareá, J.M. The Identification of ATP-Citrate Lyase as a Protein Kinase B (Akt) Substrate in Primary Adipocytes. J. Biol. Chem. 2002, 277, 33895–33900.
- McReynolds, M.R.; Chellappa, K.; Chiles, E.; Jankowski, C.; Shen, Y.; Chen, L.; Descamps, H.C.; Mukherjee, S.; Bhat, Y.R.; Lingala, S.R.; et al. NAD+ Flux Is Maintained in Aged Mice despite Lower Tissue Concentrations. Cell Syst. 2021, 12, 1160–1172.e4.
- Veech, R.L.; Eggleston, L.V.; Krebs, H.A. The Redox State of Free Nicotinamide–Adenine Dinucleotide Phosphate in the Cytoplasm of Rat Liver. Biochem. J. 1969, 115, 609–619.
- Merrill, D.K.; Guynn, R.W. The Calculation of the Cytoplasmic Free / Ratio in Brain: Effect of Electroconvulsive Seizure. Brain Res. 1981, 221, 307–318.
- Subrahmanian, N.; LaVoie, M.J. Is There a Special Relationship between Complex I Activity and Nigral Neuronal Loss in Parkinson’s Disease? A Critical Reappraisal. Brain Res. 2021, 1767, 147434.
- Borst, P. The Malate–Aspartate Shuttle (Borst Cycle): How It Started and Developed into a Major Metabolic Pathway. IUBMB Life 2020, 72, 2241–2259.

