Dysfunctional mitochondrial quality control (MQC) is implicated in the pathogenesis of Parkinson’s disease (PD). The improper selection of mitochondria for mitophagy increases reactive oxygen species (ROS) levels and lowers ATP levels. The downstream effects include oxidative damage, failure to maintain proteostasis and ion gradients, and decreased NAD+ and NADPH levels, resulting in insufficient energy metabolism and neurotransmitter synthesis. A ketosis-based metabolic therapy that increases the levels of (R)-3-hydroxybutyrate (BHB) may reverse the dysfunctional MQC by partially replacing glucose as an energy source, by stimulating mitophagy, and by decreasing inflammation. Fasting can potentially raise cytoplasmic NADPH levels by increasing the mitochondrial export and cytoplasmic metabolism of ketone body-derived citrate that increases flux through isocitrate dehydrogenase 1 (IDH1). NADPH is an essential cofactor for nitric oxide synthase, and the nitric oxide synthesized can diffuse into the mitochondrial matrix and react with electron transport chain-synthesized superoxide to form peroxynitrite. Excessive superoxide and peroxynitrite production can cause the opening of the mitochondrial permeability transition pore (mPTP) to depolarize the mitochondria and activate PINK1-dependent mitophagy. Both fasting and exercise increase ketogenesis and increase the cellular NAD+/NADH ratio, both of which are beneficial for neuronal metabolism.
- Parkinson’s disease
- mitochondrial quality control
- NADPH
- NAD
- Superoxide sentinel theory
- fasting
- exercise
- circadian
- ketone
- beta-hydroxybutyrate
1. Targeting MQC in PD
2. Interventional Strategies to Correct Dysfunctional MQC and Downstream Processes
2.1. NADPH Is Required for Proper MQC
Neuroinflammation is induced in the PD brain and proinflammatory cytokines are secreted by astrocytes and microglia leading to the expression of inducible nitric oxide synthase (iNOS) in these cell types [15][6]. iNOS synthesizes nitric oxide (•NO), which reacts with O2•− to form peroxynitrite (ONOO−). It is hypothesized that low-quality damaged mitochondria that need to be degraded through mitophagy synthesize excess O2•−, which performs a sentinel function. ONOO− is toxic and can be further metabolized into other forms of ROS/RNS that the mitochondrion is not well-equipped to detoxify. NADPH provides electrons to many of the mitochondrial antioxidants that detoxify ROS/RNS, including glutathione and thioredoxin, that further donate electrons to peroxiredoxins, glutaredoxins, vitamin C, vitamin E, and R-lipoic acid (Figure 1A).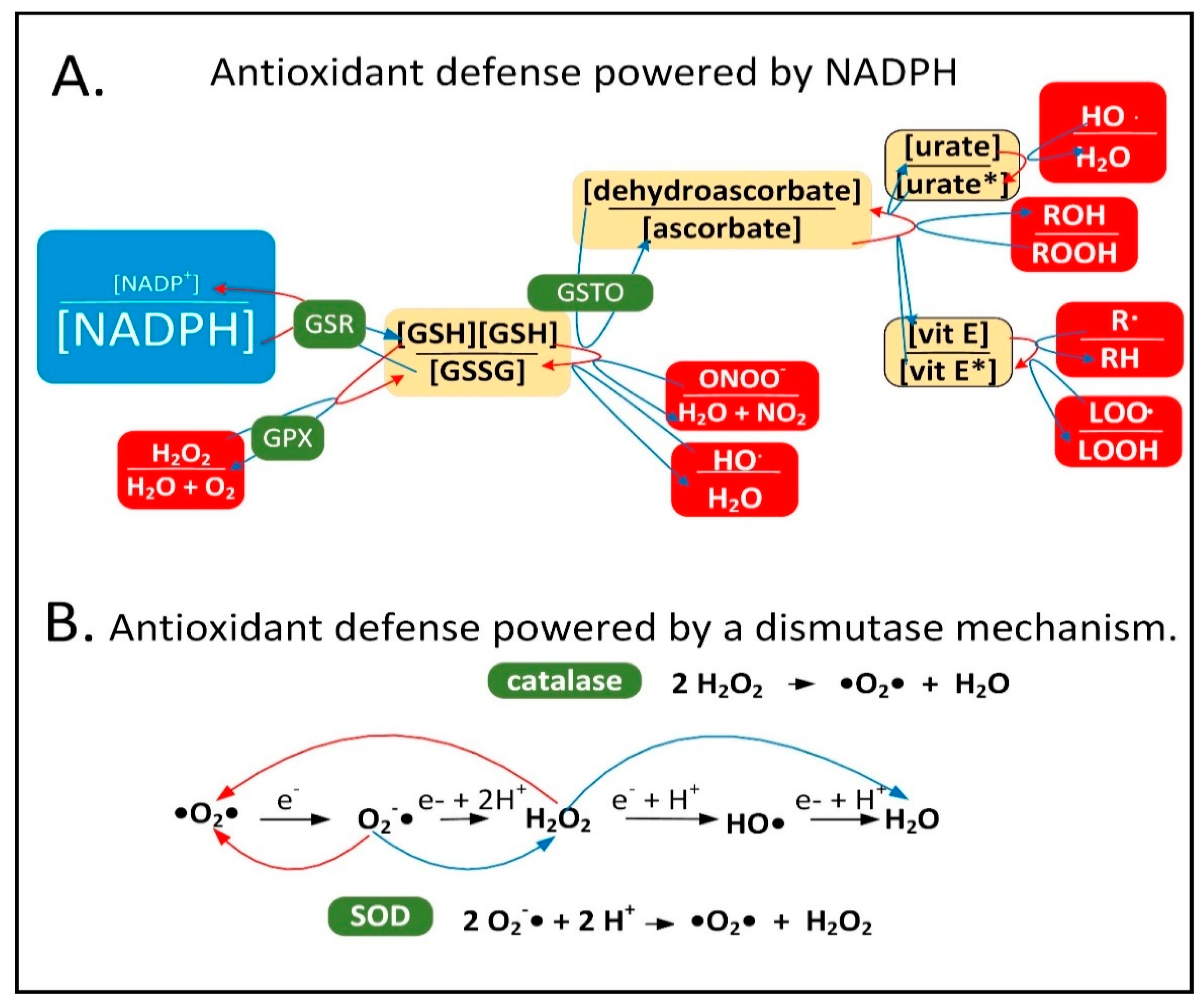
2.2. Mechanisms of Mitochondrial Protein Turnover
To maintain mitochondrial mass due to the ongoing mitophagy of organelles, there is an ongoing synthesis of mitochondrial proteins, DNA, and phospholipids. This process of mitochondrial biogenesis is largely triggered by the PGC-1 family of transcriptional co-activators including PGC-1α, PGC-1ß, and PPRC1 (PRC), and several transcription factors including NRF1 (nuclear respiratory factor 1), NRF2 (dimer of GABPA and GABPB1), ESRRA (ERR-α), ESRRG (ERR-ɣ), and NFE2L2 (Nrf2). Mitophagy in a dopaminergic neuron is complicated by the fact that the neurons have arbors of axons and dendrites that, when added together total roughly 4.1 m. Mitochondria are transported from the cell bodies down the axons to synaptic terminals to supply ATP locally at the synapses. Most mitophagy occurs in cell bodies, although some occurs in axons. Therefore, dysfunctional mitochondria at synapses are most frequently transported back to the cell body for degradation. It is estimated that there are two million mitochondria in a single dopaminergic neuron [28][11]. Therefore, the key to MQC in neurons is not only the ability to select the dysfunctional mitochondria for degradation, but also the ability to transport the dysfunctional mitochondria to the mitophagy machinery where the degradation takes place. Both processes are often dysfunctional in PD [29][12]. The regulation of MQC can be complex and for this reason the mechanisms through which individual organelles are selected for mitophagy are not that well characterized, although there is strong evidence that decreased mitochondrial membrane potential plays a role. There are several different proteolytic systems present in mitochondria including proteases inside the mitochondrial matrix (LONP1, CLPP/CLPX, PITRM1, XPNPEP3, and MIPEP), in the inner membrane (AFG3L2, SPG7, IMMP1L, IMMP2L, OMA1, PARL, YME1L1, and PMPCA/PMPCB), in the intermembrane space (HTRA2 and LACTB), and in the outer membrane (USP30) [30][13] that degrade proteins independently of mitophagy. Mitochondrial membranes that contain soluble proteins can also bud off from the mitochondria, and these are called mitochondrial-derived vesicles (MDVs). PINK1 and Parkin may be involved in the formation of MDVs. The cell may also possess a non-selective form of autophagy that slowly degrades the mitochondria indiscriminately [31][14].2.3. DJ-1 Is Regulated by Cytoplasmic NADPH and Glutathione and Controls Nrf2 Activation in Astrocytes
There is a rare human DJ-1 mutant variant linked with recessive PD. There is evidence that preventing the decline of the cytoplasmic NADPH levels in PD will preserve the function of DJ-1, a cytoplasmic redox-sensitive chaperone protein sensor of H2O2 levels and an activator of Nrf2. DJ-1 protein is present at higher levels in human astrocytes than neurons [32][15]. But unexpectedly, DJ-1 mRNA is found at higher levels in neurons than astrocytes. This suggests complex post-transcriptional regulation of DJ-1 levels and that DJ-1 could be degraded in response to high levels of neuronal H2O2. Nrf2 is a master transcriptional regulator that binds to antioxidant response elements (AREs) in the promoters of genes to induce transcription. In the brain, like DJ-1, Nrf2 is present at much higher levels in astrocytes than neurons. Complexly, DJ-1 is activated by H2O2 oxidation of cysteine 106 sulfhydryl, but too much oxidation converts this cysteine sulfhydryl into a sulfonic acid, permanently deactivating the protein and marking it for ubiquitination and proteasome degradation. NADPH and glutathione are critical in preserving the active sulfenic acid and sulfinic acid forms of DJ-1 and preventing them from being further oxidized to the inactive sulfonic acid form (Figure 2).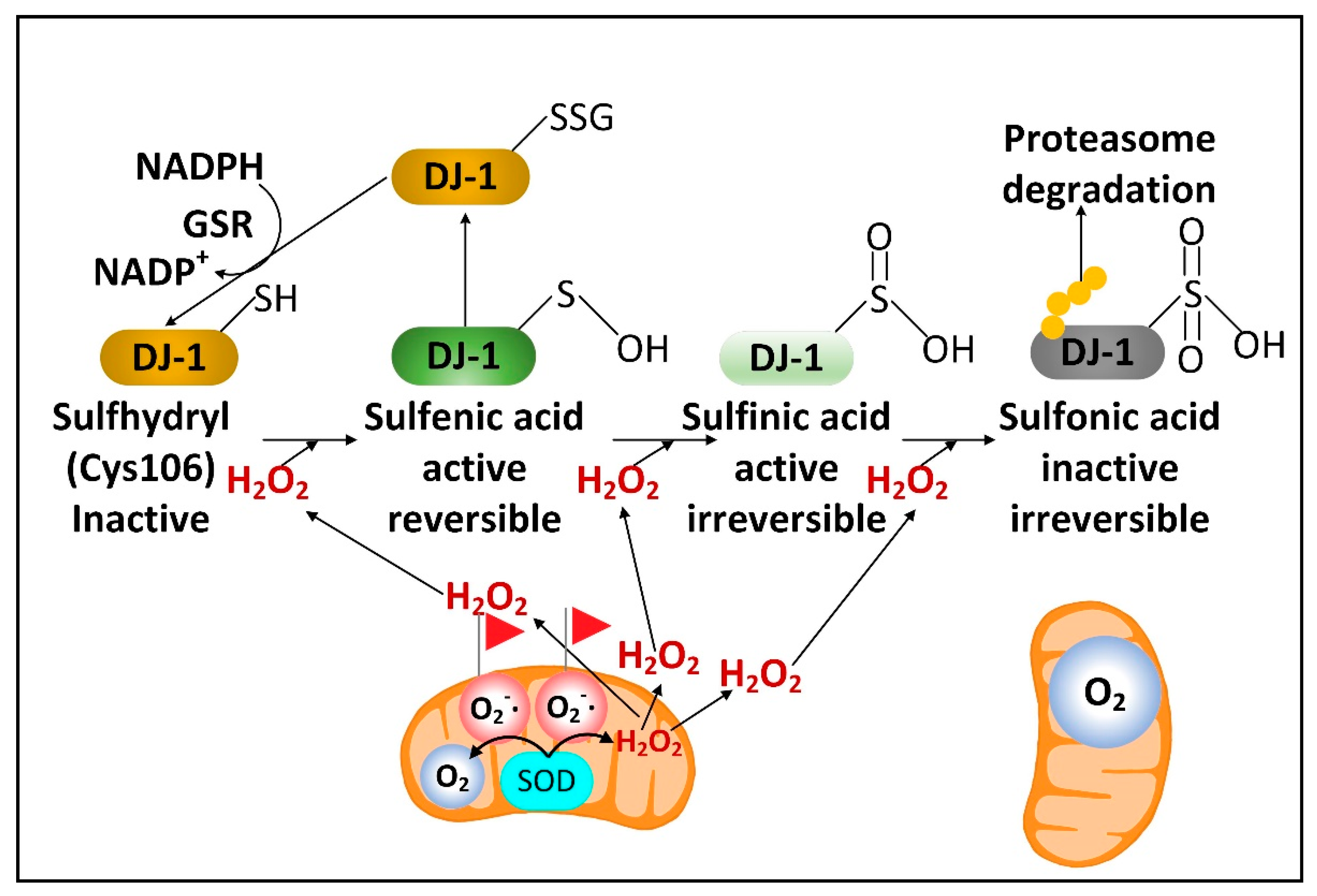
2.4. Fasting Increases the Hepatic Levels of ATF4 to Increase NADPH, FGF21, and Parkin
2.5. Fasting Activates Glutamate Dehydrogenase to Increase the Levels of the Anti-Aging Metabolite αkg
In hepatocytes and pancreatic beta-cells, fasting or CR has been shown to decrease the levels of mitochondrial sirtuin SIRT4 [59,60][42][43]. SIRT4 ADP-ribosylates and inhibits mitochondrial glutamate dehydrogenase, an enzyme which deaminates glutamate into αkg and ammonia. Therefore, under well-fed conditions when glutamate dehydrogenase activity is low, glutamate and glutamine levels increase leading to the activation of mTORC1. Under fasting conditions when glutamate dehydrogenase activity is high, αkg levels rise [60][43]. This regulation might be partly responsible for the diurnal fluctuations in skeletal muscle αkg levels [61][44]. Supplementing with αkg has been shown to increase the lifespan of C. elegans [62][45], Drosophila [63][46], and mice [64][47], while plasma αkg levels decline substantially with aging in mammals [65][48]. Brain αkg and citrate levels declined in aged rats. This decline as well as the decreased cerebral blood flow that occurred with aging were prevented by CR [66][49].2.6. Fasting Decreases the Cytoplasmic [NADP
+
]/[NADPH] Ratio in Liver, but Its Effects on Neural Cell [NADP
+
]/[NADPH] Ratios Are Not Yet Known
Flux through enzymes that synthesize cytoplasmic NADPH changes between the well-fed and fasted/ketotic states. In the well-fed state when blood and tissue glucose levels are high, the PPP is primarily used in most tissues for the synthesis of NADPH. However, after 24 h of fasting when glycogen levels are depleted human blood glucose levels start to decline. On average, blood glucose levels rose by roughly 8% following a meal and returned to baseline three to four hours after the mealtime [73][50]. Blood glucose levels in human adults were shown to have declined by 6% following a 24 h fast [74][51] and by 25% following a 72 h fast and remained stable at that level for weeks of starvation [75][52]. In comparison, adult human blood BHB levels rose to approximately 0.4 mM within 16–24 h of fasting, to 1 mM after 48 h of fasting, and to 2 mM after 72 h of fasting [76][53]. Due to the fasting-induced decline in blood and cellular glucose levels and cellular PPP flux, the PPP, together with IDH1 and ME1 (and serine catabolism in the liver), are likely used for cytoplasmic NADPH synthesis in the fasted state. Any fasting and BHB-induced boost in cytoplasmic NADPH synthesis in neural cells may be limited by the moderately low activity of IDH1 in most neural cell types [77][54] and by the lack of expression of ME1 in neurons as shown by studies of bovine brain [78][55]. As a minor source of cytoplasmic NADPH in most tissues, reducing equivalents from mitochondrial NADPH, such as those generated by NNT activity or serine and glycine catabolism, can be shuttled to the cytoplasm via the reductive decarboxylation of αkg to isocitrate catalyzed by the Krebs cycle enzyme isocitrate dehydrogenase 2 (IDH2). The IDH2-catalyzed reductive carboxylation reaction concurrently oxidizes NADPH. Next, mitochondrial isocitrate is exported to the cytoplasm, followed by the cytoplasmic IDH1-catalyzed reduction of NADP+ to NADPH linked to the oxidation of isocitrate to αkg. The decreased level of serine found in the PD subject plasma [79][56] may be a result of its increased catabolism fueling NADPH synthesis for antioxidant defense as well as its decreased synthesis caused by lowered levels of DJ-1 and ATF4 as mentioned earlier.2.7. Fasting May Improve PD Symptoms by Decreasing the Number of Senescent Astrocytes and Microglia without Affecting Substantia Nigra Mitochondrial DNA Deletion Levels
There is now substantial evidence suggesting that the accumulation of senescent cells plays a role in aging and aging-related disorders. Studies with mice demonstrated the accumulation of senescent astrocytes and microglia following the injection of alpha-synuclein pre-formed fibrils into the brain. MPP+ was also shown to stimulate cellular senescence [87][57]. Excitingly, paraquat-induced PD symptoms in mice were mitigated by killing senescent cells [88][58]. Cellular senescence in vitro is typically characterized by increased mitochondrial fusion and decreased mitophagy leading to increased mitochondrial mass and increased ROS levels [89][59]. One mechanism through which cellular senescence may be induced is by the release of mitochondrial DNA (mtDNA) into the cytoplasm by the opening of the mPTP activating the cyclic GMP-AMP synthase (cGAS) and stimulator of the interferon genes (STING) pathway [90][60]. The mPTP has a pore diameter of roughly 3 nm. Thus, linear double-strand mtDNA with a diameter of 2.3 nm appears to be threaded through the pore and released into the cytoplasm. Mitochondrial FEN1 nuclease has been shown to be activated by oxidative stress and to hydrolyze oxidized mtDNA into 500–650 base pair fragments released through the mPTP into the intermembrane space [91][61]. Mitochondrial outer membrane VDAC pores can also transport solutes such as double-strand DNA with a diameter less than 3 nm between the intermembrane space and the cytoplasm [92][62]. Although there are currently little data to support this mechanism, mitochondrial depolarization activating calcium-independent phospholipase A2-γ (PNPLA8) [93][63] may lead to the hydrolysis of mitochondrial inner membrane phospholipids to release larger mtDNA fragments. Current data suggest that PNPLA8 activated by mitochondrial depolarization hydrolyzes outer membrane phospholipids for the release of cytochrome c into the cytoplasm. MtDNA fragments of sizes up to 1000 base pairs [94,95][64][65] have been shown to be released and trigger inflammation by activating the NLRP3 inflammasome, which can contribute to cellular senescence. Supplementation with nicotinamide riboside, an NAD+ precursor, was recently demonstrated to increase mitophagy, decreasing NLRP3 inflammasome activation and cGAS-STING activity to decrease neuroinflammation and cellular senescence in an APP/PS1 mouse model of AD [96][66].2.8. A Requirement for Fasting in Dietary Restriction-Induced Longevity
Recent studies using mice have shown that dietary restriction did not extend longevity when the mice were fed many small meals without an extended period of fasting [111][67], and that 13 h daily fasting extended the mouse lifespan even when the mice were not calorie restricted [112][68]. Insulin signaling leads to the phosphorylation and exclusion from the nucleus of the pro-longevity transcription factor FOXO3A. When insulin levels decline during fasting, FOXO3A is not phosphorylated, leading to its translocation into the nucleus to mediate the changes in gene expression required for the lifespan extension that occurs due to dietary restriction/intermittent fasting [113][69]. Therefore, it appears likely that BHB mediates some of its neuroprotective and disease-modifying effects through stimulating FOXO3A-dependent transcription. Consistent with this hypothesis, the FOXO3A homolog DAF-16 is required for BHB-mediated lifespan extension in C. elegans [114][70].2.9. Evidence That Combining Fasting and Exercise Is Neuroprotective
For aIn extensive discussion of how fasting and exercise improve neural function[71], the following review article ischolars highly recommended [115]. The authors provide evidence that intermittent metabolic switching (IMS) between glucose and ketone body metabolism maintains brain function at a high level. Exercise, which stimulates glucose consumption and glycogen breakdown, facilitates the switch from glucose to ketone body metabolism. Although exercise by itself has been shown to have little effect on the improvement of the gait in PD patients [116[72][73],117], exercise has been shown to improve aspects of memory and decrease depression and anxiety in both humans and animal models [118][74]. Like fasting, exercise decreases neuroinflammation [129][75], which may be partly due to BHB binding the HCAR2 receptor and the NLRP3 inflammasome blocking proinflammatory signaling [130][76]. However, BHB administration was unable to block the NLRP3 inflammasome activated by alpha-synuclein in microglia. Encouragingly, a two-year trial of cognitive training, diet, and exercise led to improved cognition in AD patients [131][77]. This suggests that it may also be possible to use combined diet and exercise therapy to slow the progression of PD.2.10. Exercise and Its Effects on PD Brain and Muscles
Exercise is categorized into aerobic (endurance) training or anaerobic (resistance) training. The intensity and volume or duration of the workout determines whether enough O2 is transported to the skeletal muscles for aerobic metabolism to occur. Aerobic and anaerobic training are both important for the release of myokines to improve systemic homeostasis and decrease inflammation. Aerobic exercise was shown to increase the levels of the myokines VEGF-A, SPARC, sestrin, SDF-1, irisin, IL-6, IL-15, BAIBA, and apelin, while anaerobic exercise increased the levels of VEGF-A, irisin, IL-6, IL-15, IGF-1, decorin, and BMP-7. Both types of exercise decreased the myostatin secretion. With aging, the secretion of VEGF-A, SPARC, sestrin, SDF-1, irisin, IL-15, IGF-1, decorin, BMP-7, BAIBA, and apelin decreased, while that of IL-6 and myostatin increased [134][78]. Therefore, exercise therapies can now be tailored to specific aging-related diseases, such as PD. Examples of different types of exercise that have been shown to be beneficial for PD include resistance training, dance, yoga, and virtual reality training [135][79]. High-intensity interval training has also recently been demonstrated to result in endurance-like adaptations in cellular efficiency. Matched-work continuous and intermittent exercise both led to acute and chronically improved insulin sensitivity and reduced glycogen utilization and lactate production [146][80]. High-intensity interval training, with the increased recruitment of type-II fibers, led to increased AMPK activity and PGC-1α expression in response to increased exercise intensity and cellular energy demand [147][81]. Further evaluation of low-volume, high-intensity interval training indicated improved mitochondrial protein content and enzyme activities. The increased mitochondrial protein content (~25%) was consistent with that reported previously following endurance training or higher intensities of high-intensity interval training [148][82]. The increased aerobic capacity matched the increased COX I (MT-CO1) and COX IV (COX4I1) complex IV subunit levels. There were also increased levels of pyruvate dehydrogenase kinase 2 (PDK2). PDK2 phosphorylates and inhibits the pyruvate dehydrogenase complex (PDC) resulting in a decreased reliance on glycolysis and increased reliance on fatty acid beta-oxidation [148][82].2.11. Greater Metabolic Changes Induced by Exercise When the Exercise Occurs Early in the Active Period of the 24 h Circadian Cycle
Several protective compounds or “exerkines” were increased more during exercise early in the active phase, including serum AMP and beta-aminoisobutyric acid (BAIBA); muscle αkg, kynurenine, and GABA; liver BAIBA, kynurenine, and kynurenate; and hypothalamic BAIBA [155][83]. There was also increased muscle GSSG/GSH that occurred when exercise was carried out in the active phase. The more oxidized cellular environment following exercise in the active phase likely allows for more mitohormesis and ROS-induced Nrf2 activation several hours later, increasing NADPH levels and correcting the deficit. The most striking finding was the much greater global increase in 2-hydroxybutyrate (α-hydroxybutyrate) levels following active-phase exercise. In the same way that the pyruvate/lactate ratio is a marker of the cytoplasmic [NAD+]/[NADH] ratio due to the high activity of lactate dehydrogenase in most tissues [156][84], the 2-ketobutyrate/2-hydroxybutyrate ratio is also a global redox marker of the cytoplasmic [NAD+]/[NADH] ratio [157][85] due to 2-ketobutyrate and 2-hydroxybutyrate also serving as substrates for lactate dehydrogenase [158][86]. The more reduced [NAD+]/[NADH] ratio directly following exercise early in the active phase is likely an important signal to induce cytoprotective gene expression and metabolic changes leading to the restoration of the normal redox state.
2.12. Entrainment of the Circadian-Regulated Production of NAD
+
in the Morning
An alternative or complementary strategy to the oral supplementation of NAD+ precursors to raise the cellular NAD+ levels is to use fasting, exercise, and the body’s own natural circadian rhythms. During the early morning (4:00 AM) in humans, the heterodimeric transcription factor composed of the proteins BMAL1 (ARNTL) and CLOCK is most active in order to transcribe the NAMPT gene in skeletal muscle [176][87]. Therefore, during the early- to mid-morning the levels of NAD+ are likely at their highest point at least in skeletal muscle. SIRT1 consumes NAD+ in deacetylation reactions and specifically removes an acetyl group from a lysine residue in the BMAL1 transcription factor to activate the heterodimer of BMAL1 and CLOCK for transcription. The period (PER1, PER2, and PER3) genes are expressed in a circadian manner in the suprachiasmatic nucleus pacemaker. The PER proteins bind to CRY (CRY1 and CRY2) proteins to form a heterodimeric transcriptional repressor that further binds to and prevents the transcriptional activity of CLOCK-BMAL1 to restore the night-time pattern of gene expression. PER2 lysine acetylation increases PER2 function, and the acetyl group is removed by SIRT1, which inhibits protein function. The sum of these processes cause NAD+ levels to be entrained by the circadian transcription factors. Therefore, the activity of the heterodimeric transcription factor CLOCK-BMAL1, the transcription of the NAMPT gene, and NAD+ levels increase in unison [177][88]. In mice NAD+ levels are altered by up to 40% during different times of the 24-h cycle [178,179][89][90]. In contrast to peak human skeletal muscle NAD+ levels that occur in the morning, the circadian oscillations in human mitochondrial respiration peak at roughly 11:00 PM, with the trough occurring at 1:00 PM [180][91]. The reason for why these different metabolic oscillations are not in phase is not well understood. In studies of humans performing roughly 14 h fasts, it was shown that fasting from dinner until breakfast led to more lipid oxidation than fasting from bedtime until lunch [181][92]. These results may be due to increased rates of fatty acid beta-oxidation during fasting and the circadian increase in ETC function in the evening. These events lead to higher rates of fatty acid beta-oxidation during evening fasts compared to those that occur during morning fasts, when diurnal ETC activity is not at its peak.2.13. Circadian Regulation of Mitophagy and Mitochondrial Dynamics May Play a Role in PD
The tyrosine hydroxylase gene, as well as dopamine receptor genes, are expressed in a circadian manner [191][93]. The decreased dopamine and melatonin levels and increased cortisol levels in PD patients are associated with decreased BMAL1 expression [192,193][94][95]. Long term L-DOPA therapy [194][96] or increased neuroinflammation [195][97] were shown to accelerate the circadian dysfunction in PD rat models. The circadian regulators BMAL1 [196][98] and REV-ERBα [197][99] have been shown to preserve motor function and reduce neuroinflammation in mouse models of PD. The role of alterations in circadian gene expression in neuroinflammation and the pathogenesis of PD has recently been reviewed [183,198]. Not only does mitochondrial ETC complex activity follow circadian rhythms [199][100], but mitophagy rates and mitochondrial dynamics also follow a diurnal pattern [200][101]. In the liver, BMAL1 induces the expression of PINK1, the mitochondrial fission protein FIS1, and the mitophagy receptor BNIP3. The expression of the mitochondrial fission protein DRP1 also fluctuates in a diurnal pattern [201][102]. The loss of the CLOCK protein disrupts the expression of the autophagy proteins ATG7, RAB7a, TFEB, and SQSTM1 [202][103]. Conversely, Parkin mutations in PD patients also disrupt circadian gene expression in iPSC-derived neurons [202][103]. HDAC3 negatively regulates the circadian clock and mitophagy by activating Rev-erbα, a negative regulator of BMAL1 [181][92], and possibly by inhibiting PGC-1α expression [203][104].3. Dopaminergic Neurons Are Vulnerable to Dysfunctional MQC
The discovery that the PD-causing toxin MPTP was converted to MPP+ by the enzyme monoamine oxidase-B (MAO-B) to cause dopaminergic neuron loss and PD symptoms led to a quest to find the mechanism underlying neuronal toxicity. MPTP was found to be taken up by astrocytes and serotonergic neurons that express MAO-B and then converted into MPP+, which was released into the extracellular space. MPP+ was then found to be taken up by dopamine transporters expressed in dopaminergic neurons [206][105]. From the cytoplasm, MPP+ is concentrated 40-fold in the mitochondrial matrix [207][106], resulting in the inhibition of ETC complex I. Therefore, the most efficient mitochondria with the highest membrane potential are the ones that accumulate the most MPP+ resulting in the most ETC inhibition [207][106]. Does the inhibition of complex I by MPP+, rotenone, or alpha-synuclein [208][107] depolarize the mitochondrial inner membrane enough to stimulate mitophagy when complex II is still active for electron transport? The answer may depend upon the cell type studied. However, even if mitochondrial depolarization does not occur immediately, it likely eventually occurs because of the increased O2•− generation that results from complex I inhibition, leading to increased ONOO− and H2O2 formation, which open the mPTP to completely depolarize the mitochondrion [209][108]. In either case, the data strongly suggest that interfering with MQC leads to PD. Therefore, PD treatments should be directed towards increasing mitochondrial resilience by improving the redox state. MPP+ concentrated in the mitochondrial matrix undergoes redox cycling [210][109], a futile oxidation-reduction cycle that generates O2•− (Figure 43) [211][110]. The redox potential for the conversion of MPP+ to MPP• radical is approximately −1.0 V. The mitochondrial or cytoplasmic [NADP+]/[NADPH] has a redox potential of only −0.42 V [170][111], and therefore, NADPH cannot reduce MPP+. Two-electron reduction pathways have a higher redox potential of approximately −0.84 V. Thus, two-electron reducing agents present in the mitochondrial matrix were sought, and two candidates, ALDH2 (aldehyde dehydrogenase H2) and DLD (dihydrolipoamide dehydrogenase), that could reduce MPP+ were identified [210,212][109][112].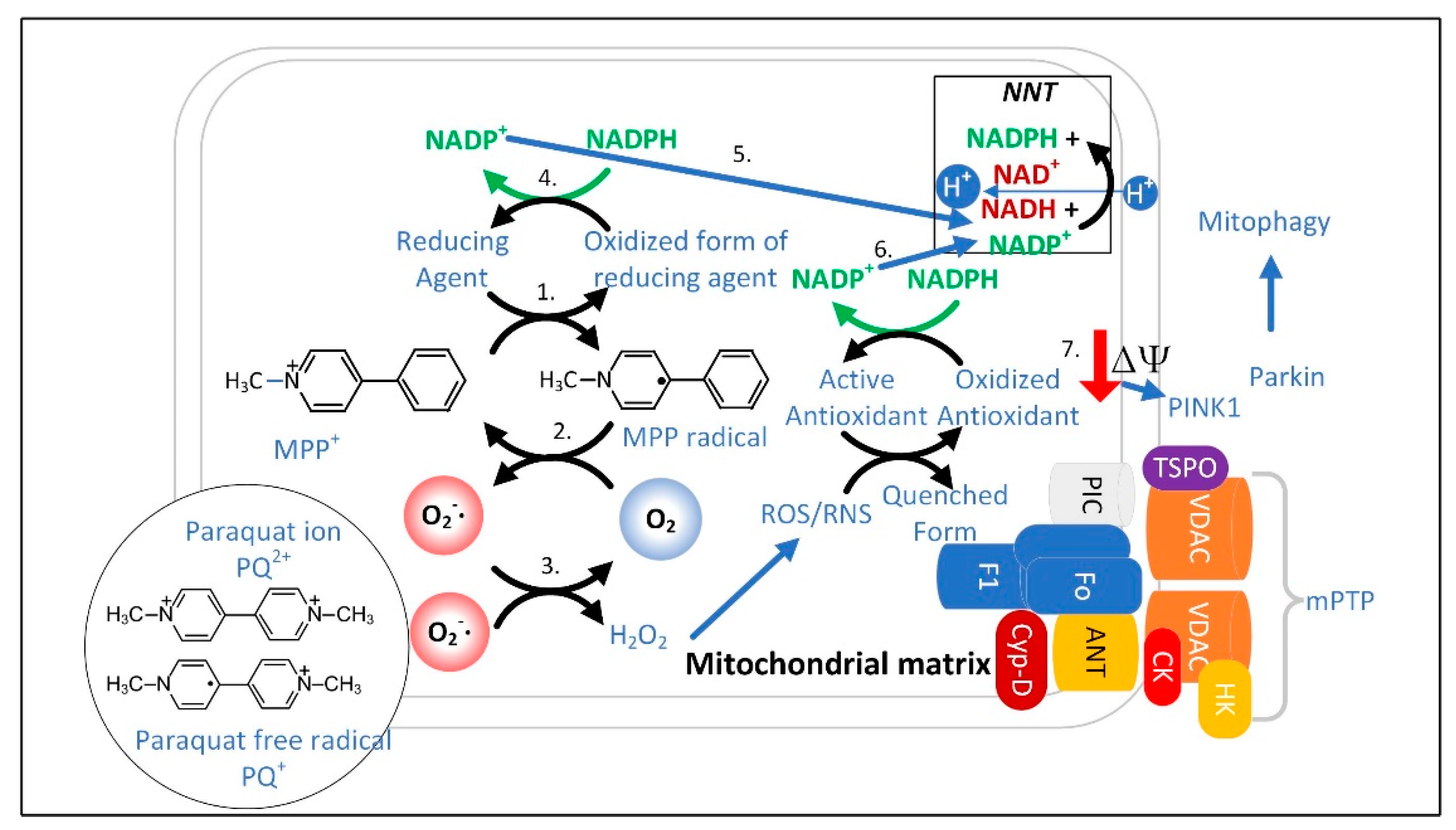
3.1. The Superoxide Sentinel Hypothesis of MQC
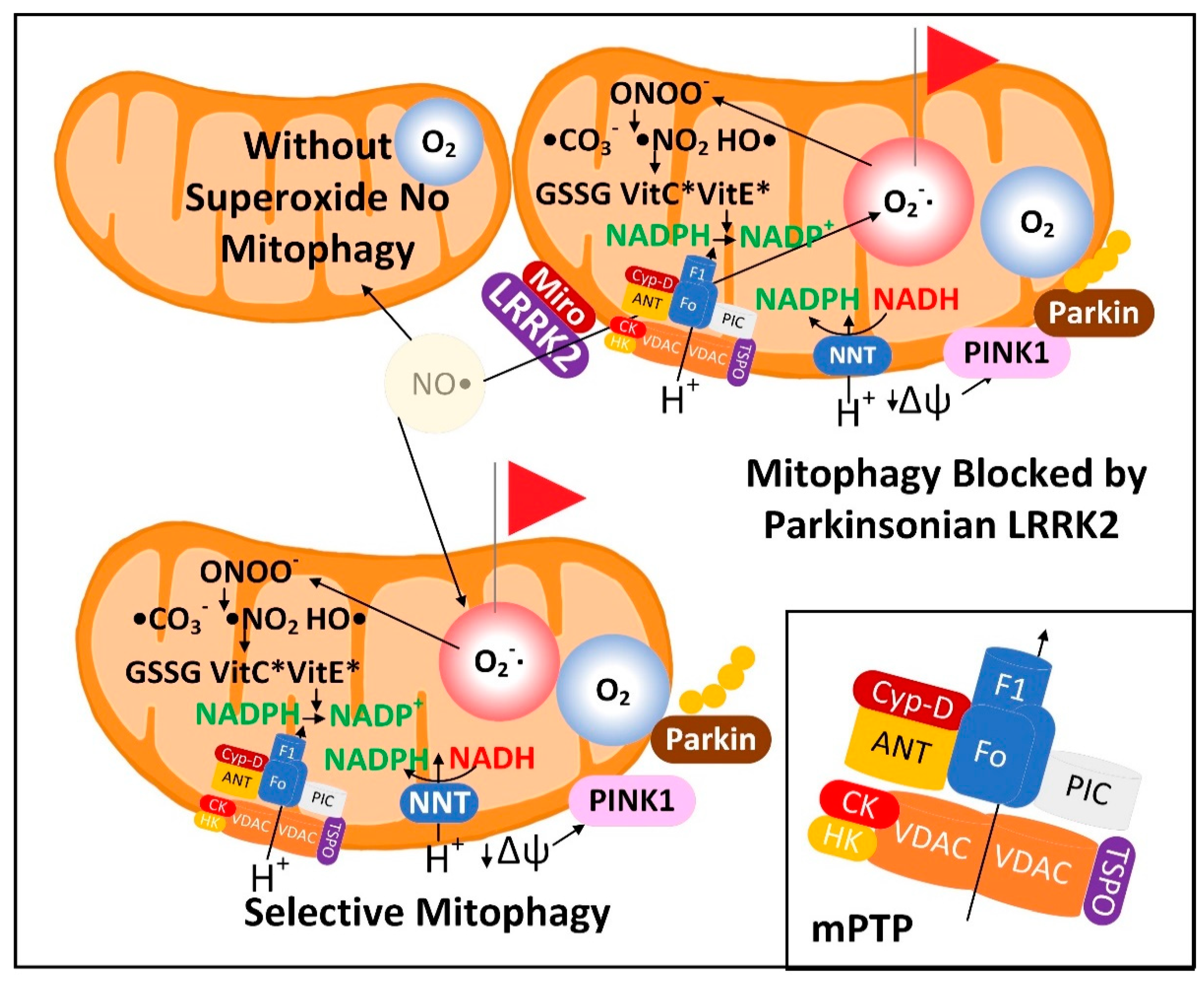
3.2. Neurons with Low-Quality Damaged Mitochondria Likely Show Decreased Proteolysis of α-Synuclein
Postmitotic cells such as neurons cannot dilute cytoplasmic contents through cell division, and so high concentrations of oxidized and aggregated proteins such as α-synuclein can potentially accumulate. Therefore, postmitotic cells rely on efficient proteolytic systems to prevent protein accumulation. The α-synuclein protein can undergo several post-translational modifications that favor aggregation and lead to toxicity [234][122]. The cell has two major proteolytic systems for clearing damaged or aggregated cytoplasmic proteins such as α-synuclein [234][122], and they both require sufficient ATP. In the ubiquitin-proteasome system, ubiquitin is covalently attached to α-synuclein or another protein that exposes a hydrophobic core on the surface of the protein. Both the post-translational modification attaching ubiquitin to the protein to be degraded and the proteolysis by the proteasome require ATP [235,236][123][124]. Autophagy is the other major proteolytic system used to degrade α-synuclein and other cytoplasmic proteins and organelles. Autophagy results in complete lysosomal proteolysis yielding individual amino acids. The various types of autophagy, including macroautophagy, microautophagy, and chaperone-mediated autophagy, all require ATP [237][125].3.3. Limited Clearance of ROS/RNS Leads to the Oxidation and Depletion of Cardiolipin and Plasmalogens
Another outcome of poor MQC is the increased oxidation of polyunsaturated fatty acids by ROS to form isoprostanes, malondialdehyde, or 4-hydroxynonenal (4-HNE), which inhibit the function of proteins. For example, IDH1 is susceptible to covalent modification and inhibition by 4-HNE [248][126]. 4-HNE is produced by •OH attacking polyunsaturated fatty acids. Limiting dietary omega-6 polyunsaturated fatty acids, such as those that are present at high levels in vegetable oils (excluding olive oil), may be beneficial in decreasing 4-HNE synthesis. Consistent with this hypothesis, high levels of the omega-6 fatty acids dihomo-gamma-linolenic acid (DGLA) [249][127] and arachidonic acid (AA) [250][128] have been shown to cause ferroptosis. PD neural cells are characterized by lipidopathy, particularly of sphingolipids. PD neurons and microglia are characterized by increased levels of fatty acids and lipid droplets, while PD astrocytes show decreased levels of these lipids [251][129]. Poor MQC also leads to the oxidation and depletion of mitochondrial cardiolipin (a diphosphatidylglyceride lipid with two phosphate groups and four acyl chains) and plasmalogens (vinyl-ether phospholipids enriched with AA and docosahexaenoic acid (DHA)). Cardiolipin constitutes roughly 20% of the inner mitochondrial membrane phospholipids and is critical for the mitochondrial inner membrane to maintain a membrane potential, as each cardiolipin molecule can bind a proton. When oxidatively damaged cardiolipin binds cytochrome c it activates a latent peroxidase [252][130] and plasmalogenase [253][131] activity, which can contribute to cell death. PLA2G6, group VIA calcium-independent phospholipase A2-beta (iPLA2-β), is a gene that when mutated causes PD, and the gene product has a partial mitochondrial localization. iPLA2-β [254][132], as well as the related mitochondrial and peroxisomal-localized iPLA2-ɣ [255][133], can hydrolyze fatty acids from cardiolipin. The depletion of cardiolipin can cause a direct inhibition of mitophagy, as the redistribution of cardiolipin from the inner mitochondrial membrane to the outer mitochondrial membrane is an important step in mitochondrial fission and mitophagy. The Drp1 protein that is recruited to the mitochondria to mediate mitochondrial fission [256][134] and the LC3/Atg8 protein involved in autophagosome formation [257][135] bind cardiolipin as important steps in MQC. Cardiolipin is also required for the activity of the NLRP3 inflammasome [258][136]. So, cells may have evolved to decrease cardiolipin levels in times of oxidative stress to reduce inflammation..4. NADPH May Decrease in PD Neurons and Glia Limiting the Synthesis of Serotonin, Melatonin, Epinephrine, Norepinephrine, and •NO
3.4. NADPH May Decrease in PD Neurons and Glia Limiting the Synthesis of Serotonin, Melatonin, Epinephrine, Norepinephrine, and •NO
The increase in the cellular [NADP+]/[NADPH] ratio in PD neural cells resulting from dysfunctional MQC and increased O2•− generation may decrease astrocyte NADPH-dependent fatty acid synthesis, thus decreasing the quantity of neutral lipids [271][137] and may also decrease the NADPH-dependent neuronal synthesis of many neurotransmitters. Due to a decline in NADPH, the active coenzyme tetrahydrobiopterin (BH4) becomes oxidized to the inactive form biopterin (BH2), and thus becomes unavailable in the quantities required for efficient synthesis of dopamine [229][138], epinephrine, and norepinephrine (Figure 75), as well as serotonin, melatonin, and •NO. Decreased mitochondrial NADPH can also limit the synthesis of proline required for protein synthesis [272][139].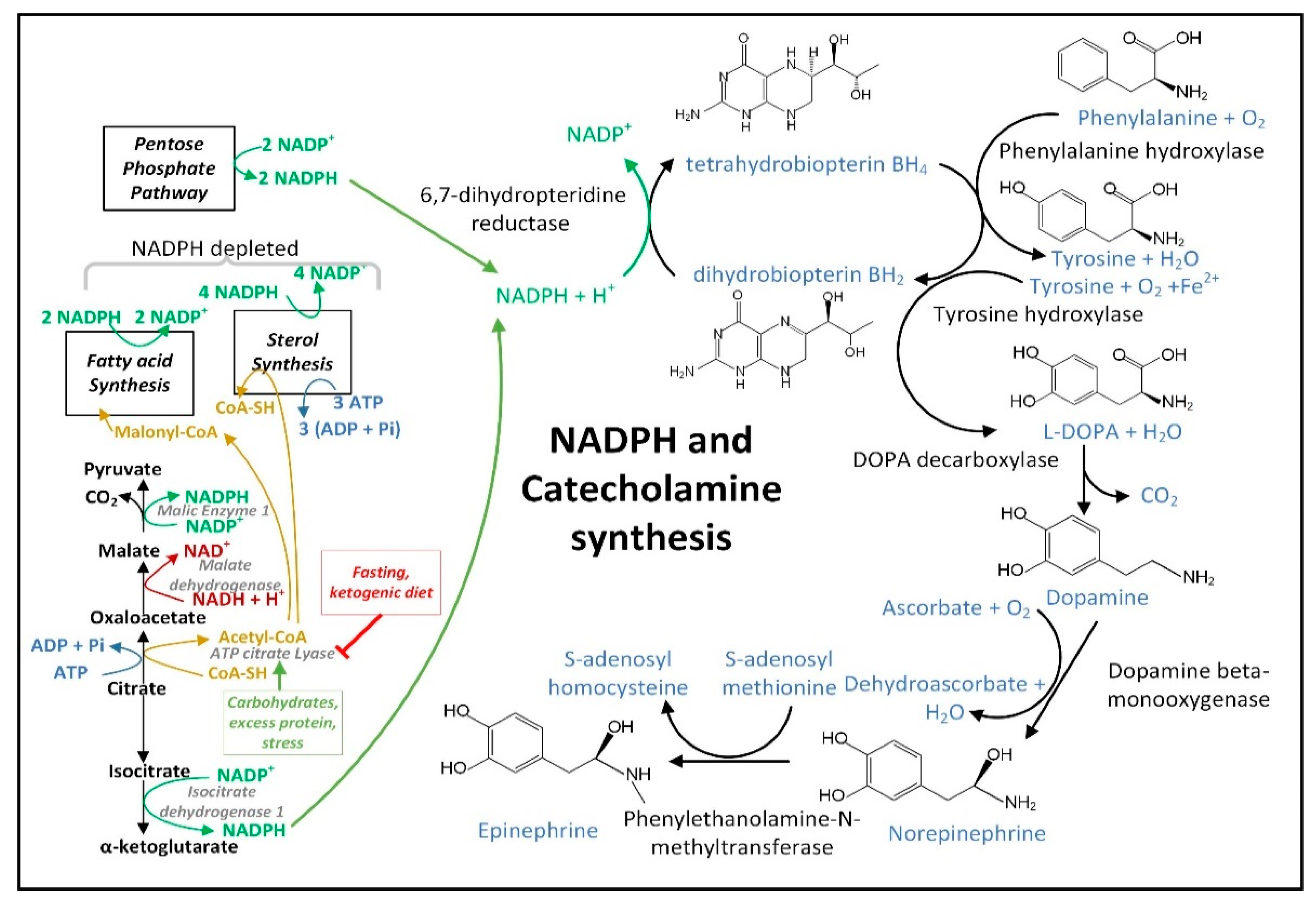
3.5. Insulin Released after Feeding Increases Cytoplasmic NADPH Oxidation by Stimulating Fatty Acid Synthesis
The decreased hepatic cytoplasmic [NADP+]/[NADPH] ratio that occurs during fasting may require glucagon [273][140] and may not occur after carbohydrate consumption in part due to insulin signaling. When insulin binds its receptor on the plasma membrane, a signaling cascade is initiated activating protein kinase AKT that phosphorylates ATP-citrate lyase (ACLY) serine 454 to activate the enzyme [274][141]. Thus, the citrate that is exported from the mitochondrial matrix is diverted away from the citrate–αkg shuttle that uses IDH1 for NADPH synthesis to boost cytoplasmic NADPH levels, and instead it is routed to the citrate–malate or citrate–pyruvate shuttles that use ACLY for the synthesis of acetyl-CoA and oxaloacetate. Acetyl-CoA is largely used for NADPH-depleting fatty acid synthesis. The malate dehydrogenase 1 (MDH1) enzyme converts the generated oxaloacetate into malate, which can either be transported back into the mitochondrial matrix for the citrate–malate shuttle or converted to pyruvate by cytoplasmic ME1 with the concurrent reduction of NADP+ to NADPH as part of the citrate–pyruvate shuttle. The pyruvate is then transported back into the mitochondrial matrix.
3.6. Experiments Are Needed to Determine the (Free) Cytoplasmic [NADP
+
]/[NADPH] Ratio in Neural Cells from Aged and PD Model Mice and How These Are Altered by Fasting
When measuring total levels of NADP+ and NADPH, it was surprisingly shown that the mouse brain NADP+/NADPH ratio decreased with aging, mostly due to increases in NADPH levels [283][142]. The brain NADH level also increased with aging. In liver and brown adipose tissue NADPH levels declined with aging as would be expected for a condition characterized by increased oxidative stress. However, aging-induced NADPH loss was not observed in most tissues. Due to its known health benefits, fasting might be expected to universally reduce the total NADP+/NADPH ratio to combat oxidative stress. But in rat heart and skeletal muscle fasting led to an oxidation of this coenzyme couple. So redox changes are tissue specific. Measurements of total pyridine nucleotide ratios are also misleading and do not reflect the relevant (free) cytoplasmic [NADP+]/[NADPH] ratio, as much more NADPH than NADP+ is bound to protein and unavailable for enzyme function. Measurements of the pyruvate/malate ratio or αkg/isocitrate ratio have been commonly used to estimate the (free) cytoplasmic [NADP+]/[NADPH] in liver [82][143]. Results from a first attempt at using these metabolite ratios from whole brain to estimate cytoplasmic [NADP+]/[NADPH] yielded a promising average value across neural cell types [284][144].3.7. Dysfunctional MQC Leads to Loss of Circadian Entrainment of NAD
+
Synthesis
Dysfunctional MQC in PD decreases oxidative phosphorylation and shifts more of the burden of ATP synthesis to glycolysis. Increased glycolysis stimulates the rate of conversion of NAD+ to NADH by the GAPDH enzyme decreasing the cytoplasmic NAD+/NADH ratio and SIRT1 activity if the combined NADH oxidizing activities of mitochondrial ETC complex I and cytoplasmic lactate dehydrogenase do not increase in parallel. The ETC complex I dysfunction in PD [289][145] decreases the mitochondrial NAD+/NADH ratio, but this reduced mitochondrial NAD+/NADH ratio is not efficiently transferred to the cytoplasm due to the irreversibility of the malate–aspartate shuttle due to the coupling of the mitochondrial glutamate import (and aspartate export) to the proton-motive force during SLC25A12 and SLC25A13 carrier function [290][146]. This irreversibility maintains the roughly 100-fold higher [NAD+]/[NADH] ratio in the cytoplasm as compared to the mitochondrial matrix. The [NAD+]/[NADH] ratio is roughly 700 in the cytoplasm and is roughly 7 in the mitochondrial matrix [156][84].4. Conclusions
MQC becomes dysfunctional in PD and is a primary driver of the onset of disease and disease progression. When performed individually, fasting and exercise have been shown to have only minimal effects in the treatment of PD. However, combining an overnight and morning fast with morning exercise (and coffee consumption) may synergize with circadian oscillations in NAD+, NADPH, and mitophagy rates and be a promising therapy for PD. It is hypothesized that the increased levels of BHB, glucagon, ghrelin, and FGF21, and the decreased levels of insulin play major roles in providing the neuroprotection in this therapy. In addition to activating Nrf2 and possibly ATF4 to induce the expression of NADPH-synthesizing enzymes, BHB also activates neuroprotective pathways, inhibits proinflammatory mediators such as the NLRP3 inflammasome, and induces epigenetic effects that likely contribute to its neuroprotective effects. Clinical trials will aid in the determination of how efficacious this therapy is across the wide spectrum of sporadic and familial PD cases.References
- Panicker, N.; Ge, P.; Dawson, V.L.; Dawson, T.M. The Cell Biology of Parkinson’s Disease. J. Cell Biol. 2021, 220, e202012095.
- Singh, F.; Ganley, I.G. Parkinson’s Disease and Mitophagy: An Emerging Role for LRRK2. Biochem. Soc. Trans. 2021, 49, 551–562.
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The Pharmacological Regulation of Cellular Mitophagy. Nat. Chem. Biol. 2017, 13, 136–146.
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211.
- Butkovich, L.M.; Houser, M.C.; Chalermpalanupap, T.; Porter-Stransky, K.A.; Iannitelli, A.F.; Boles, J.S.; Lloyd, G.M.; Coomes, A.S.; Eidson, L.N.; De Sousa Rodrigues, M.E.; et al. Transgenic Mice Expressing Human α-Synuclein in Noradrenergic Neurons Develop Locus Ceruleus Pathology and Nonmotor Features of Parkinson’s Disease. J. Neurosci. 2020, 40, 7559–7576.
- McCarty, M.F.; Lerner, A. Nutraceuticals Targeting Generation and Oxidant Activity of Peroxynitrite May Aid Prevention and Control of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3624.
- Rodriguez-Enriquez, S.; He, L.; Lemasters, J.J. Role of Mitochondrial Permeability Transition Pores in Mitochondrial Autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2463–2472.
- Lemasters, J.J.; Nieminen, A.-L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The Mitochondrial Permeability Transition in Cell Death: A Common Mechanism in Necrosis, Apoptosis and Autophagy. Biochim. Biophys. Acta Bioenerg. 1998, 1366, 177–196.
- Salvi, M.; Battaglia, V.; Brunati, A.M.; Rocca, N.L.; Tibaldi, E.; Pietrangeli, P.; Marcocci, L.; Mondovi, B.; Rossi, C.A.; Toninello, A. Catalase Takes Part in Rat Liver Mitochondria Oxidative Stress Defense. J. Biol. Chem. 2007, 282, 24407–24415.
- Radi, R.; Turrens, J.F.; Chang, L.Y.; Bush, K.M.; Crapo, J.D.; Freeman, B.A. Detection of Catalase in Rat Heart Mitochondria. J. Biol. Chem. 1991, 266, 22028–22034.
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666.
- Lamberts, J.T.; Hildebrandt, E.N.; Brundin, P. Spreading of α-Synuclein in the Face of Axonal Transport Deficits in Parkinson’s Disease: A Speculative Synthesis. Neurobiol. Dis. 2015, 77, 276–283.
- Gomez-Fabra Gala, M.; Vögtle, F. Mitochondrial Proteases in Human Diseases. FEBS Lett. 2021, 595, 1205–1222.
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide Is the Major Reactive Oxygen Species Regulating Autophagy. Cell Death Differ. 2009, 16, 1040–1052.
- Bandopadhyay, R.; Kingsbury, A.E.; Cookson, M.R.; Reid, A.R.; Evans, I.M.; Hope, A.D.; Pittman, A.M.; Lashley, T.; Canet-Aviles, R.; Miller, D.W.; et al. The Expression of DJ-1 (PARK7) in Normal Human CNS and Idiopathic Parkinson’s Disease. Brain 2004, 127, 420–430.
- Blackinton, J.; Lakshminarasimhan, M.; Thomas, K.J.; Ahmad, R.; Greggio, E.; Raza, A.S.; Cookson, M.R.; Wilson, M.A. Formation of a Stabilized Cysteine Sulfinic Acid Is Critical for the Mitochondrial Function of the Parkinsonism Protein DJ-1. J. Biol. Chem. 2009, 284, 6476–6485.
- Holmström, K.M.; Kostov, R.V.; Dinkova-Kostova, A.T. The Multifaceted Role of Nrf2 in Mitochondrial Function. Curr. Opin. Toxicol. 2016, 1, 80–91.
- Yao, W.; Lin, S.; Su, J.; Cao, Q.; Chen, Y.; Chen, J.; Zhang, Z.; Hashimoto, K.; Qi, Q.; Zhang, J. Activation of BDNF by Transcription Factor Nrf2 Contributes to Antidepressant-like Actions in Rodents. Transl. Psychiatry 2021, 11, 140.
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the Mechanism of Pathogenesis of Parkinson’s Disease. J. Bioenerg. Biomembr. 2019, 51, 175–188.
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant Stress Evoked by Pacemaking in Dopaminergic Neurons Is Attenuated by DJ-1. Nature 2010, 468, 696–700.
- Xu, S.; Yang, X.; Qian, Y.; Xiao, Q. Parkinson’s Disease-Related DJ-1 Modulates the Expression of Uncoupling Protein 4 against Oxidative Stress. J. Neurochem. 2018, 145, 312–322.
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.K.; Alam, J. Identification of Activating Transcription Factor 4 (ATF4) as an Nrf2-Interacting Protein: Implication for Heme Oxygenase-1 Gene Regulation. J. Biol. Chem. 2001, 276, 20858–20865.
- Zong, Z.-H.; Du, Z.-X.; Li, N.; Li, C.; Zhang, Q.; Liu, B.-Q.; Guan, Y.; Wang, H.-Q. Implication of Nrf2 and ATF4 in Differential Induction of CHOP by Proteasome Inhibition in Thyroid Cancer Cells. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1395–1404.
- Almeida, L.M.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. The PERKs of Mitochondria Protection during Stress: Insights for PERK Modulation in Neurodegenerative and Metabolic Diseases. Biol. Rev. Camb. Philos. Soc. 2022; early view.
- Lange, P.S.; Chavez, J.C.; Pinto, J.T.; Coppola, G.; Sun, C.-W.; Townes, T.M.; Geschwind, D.H.; Ratan, R.R. ATF4 Is an Oxidative Stress–Inducible, Prodeath Transcription Factor in Neurons in Vitro and in Vivo. J. Exp. Med. 2008, 205, 1227–1242.
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-Omics Analysis Identifies ATF4 as a Key Regulator of the Mitochondrial Stress Response in Mammals. J. Cell Biol. 2017, 216, 2027–2045.
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 Pathway and Its Cross Talk with Nrf2 in Mitochondrial Quality Control. J. Clin. Biochem. Nutr. 2019, 64, 18–37.
- Torrence, M.E.; MacArthur, M.R.; Hosios, A.M.; Valvezan, A.J.; Asara, J.M.; Mitchell, J.R.; Manning, B.D. The MTORC1-Mediated Activation of ATF4 Promotes Protein and Glutathione Synthesis Downstream of Growth Signals. eLife 2021, 10, e63326.
- Renz, P.F.; Valdivia-Francia, F.; Sendoel, A. Some like It Translated: Small ORFs in the 5′UTR. Exp. Cell Res. 2020, 396, 112229.
- Endo, J.; Sano, M.; Katayama, T.; Hishiki, T.; Shinmura, K.; Morizane, S.; Matsuhashi, T.; Katsumata, Y.; Zhang, Y.; Ito, H.; et al. Metabolic Remodeling Induced by Mitochondrial Aldehyde Stress Stimulates Tolerance to Oxidative Stress in the Heart. Circ. Res. 2009, 105, 1118–1127.
- Wang, X.; Zhang, G.; Dasgupta, S.; Niewold, E.L.; Li, C.; Li, Q.; Luo, X.; Tan, L.; Ferdous, A.; Lorenzi, P.L.; et al. ATF4 Protects the Heart From Failure by Antagonizing Oxidative Stress. Circ. Res. 2022, 131, 91–105.
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. MTORC1 Induces Purine Synthesis Through Control of the Mitochondrial Tetrahydrofolate Cycle. Science 2016, 351, 728–733.
- Hill, C.M.; Albarado, D.C.; Coco, L.G.; Spann, R.A.; Khan, M.S.; Qualls-Creekmore, E.; Burk, D.H.; Burke, S.J.; Collier, J.J.; Yu, S.; et al. FGF21 Is Required for Protein Restriction to Extend Lifespan and Improve Metabolic Health in Male Mice. Nat. Commun. 2022, 13, 1897.
- Zhang, Y.; Xie, Y.; Berglund, E.D.; Coate, K.C.; He, T.T.; Katafuchi, T.; Xiao, G.; Potthoff, M.J.; Wei, W.; Wan, Y.; et al. The Starvation Hormone, Fibroblast Growth Factor-21, Extends Lifespan in Mice. eLife 2012, 1, e00065.
- Klaus, S.; Igual Gil, C.; Ost, M. Regulation of Diurnal Energy Balance by Mitokines. Cell Mol. Life Sci. 2021, 78, 3369–3384.
- Kim, K.H.; Kim, S.H.; Min, Y.-K.; Yang, H.-M.; Lee, J.-B.; Lee, M.-S. Acute Exercise Induces FGF21 Expression in Mice and in Healthy Humans. PLoS ONE 2013, 8, e63517.
- Wan, X.; Lu, X.; Xiao, Y.; Lin, Y.; Zhu, H.; Ding, T.; Yang, Y.; Huang, Y.; Zhang, Y.; Liu, Y.-L.; et al. ATF4- and CHOP-Dependent Induction of FGF21 through Endoplasmic Reticulum Stress. BioMed Res. Int. 2014, 2014, e807874.
- Bouman, L.; Schlierf, A.; Lutz, A.K.; Shan, J.; Deinlein, A.; Kast, J.; Galehdar, Z.; Palmisano, V.; Patenge, N.; Berg, D.; et al. Parkin Is Transcriptionally Regulated by ATF4: Evidence for an Interconnection between Mitochondrial Stress and ER Stress. Cell Death. Differ. 2011, 18, 769–782.
- Li, W.; Li, X.; Miller, R.A. ATF4 Activity: A Common Feature Shared by Many Kinds of Slow-Aging Mice. Aging Cell 2014, 13, 1012–1018.
- Koyanagi, S.; Hamdan, A.M.; Horiguchi, M.; Kusunose, N.; Okamoto, A.; Matsunaga, N.; Ohdo, S. CAMP-Response Element (CRE)-Mediated Transcription by Activating Transcription Factor-4 (ATF4) Is Essential for Circadian Expression of the Period2 Gene. J. Biol. Chem. 2011, 286, 32416–32423.
- Pathak, S.S.; Liu, D.; Li, T.; de Zavalia, N.; Zhu, L.; Li, J.; Karthikeyan, R.; Alain, T.; Liu, A.C.; Storch, K.-F.; et al. The EIF2α Kinase GCN2 Modulates Period and Rhythmicity of the Circadian Clock by Translational Control of Atf4. Neuron 2019, 104, 724–735.e6.
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic β Cells. Cell 2006, 126, 941–954.
- Shaw, E.; Talwadekar, M.; Rashida, Z.; Mohan, N.; Acharya, A.; Khatri, S.; Laxman, S.; Kolthur-Seetharam, U. Anabolic SIRT4 Exerts Retrograde Control over TORC1 Signaling by Glutamine Sparing in the Mitochondria. Mol. Cell Biol. 2020, 40, e00212-19.
- de Goede, P.; Wüst, R.C.I.; Schomakers, B.V.; Denis, S.; Vaz, F.M.; Pras-Raves, M.L.; van Weeghel, M.; Yi, C.-X.; Kalsbeek, A.; Houtkooper, R.H. Time-Restricted Feeding during the Inactive Phase Abolishes the Daily Rhythm in Mitochondrial Respiration in Rat Skeletal Muscle. FASEB J. 2022, 36, e22133.
- Chin, R.M.; Fu, X.; Pai, M.Y.; Vergnes, L.; Hwang, H.; Deng, G.; Diep, S.; Lomenick, B.; Meli, V.S.; Monsalve, G.C.; et al. The Metabolite Alpha-Ketoglutarate Extends Lifespan by Inhibiting the ATP Synthase and TOR. Nature 2014, 510, 397–401.
- Su, Y.; Wang, T.; Wu, N.; Li, D.; Fan, X.; Xu, Z.; Mishra, S.K.; Yang, M. Alpha-Ketoglutarate Extends Drosophila Lifespan by Inhibiting MTOR and Activating AMPK. Aging 2019, 11, 4183–4197.
- Shahmirzadi, A.A.; Edgar, D.; Liao, C.-Y.; Hsu, Y.-M.; Lucanic, M.; Shahmirzadi, A.A.; Wiley, C.D.; Gan, G.; Kim, D.E.; Kasler, H.G.; et al. Alpha-Ketoglutarate, an Endogenous Metabolite, Extends Lifespan and Compresses Morbidity in Aging Mice. Cell Metab. 2020, 32, 447–456.e6.
- Harrison, A.P.; Pierzynowski, S.G. Biological Effects of 2-Oxoglutarate with Particular Emphasis on the Regulation of Protein, Mineral and Lipid Absorption/Metabolism, Muscle Performance, Kidney Function, Bone Formation and Cancerogenesis, All Viewed from a Healthy Ageing Perspective State of the Art—Review Article. J. Physiol. Pharmacol. 2008, 59, 91–106.
- Lin, A.-L.; Zhang, W.; Gao, X.; Watts, L. Caloric Restriction Increases Ketone Bodies Metabolism and Preserves Blood Flow in Aging Brain. Neurobiol. Aging 2015, 36, 2296–2303.
- Moebus, S.; Göres, L.; Lösch, C.; Jöckel, K.-H. Impact of Time since Last Caloric Intake on Blood Glucose Levels. Eur. J. Epidemiol. 2011, 26, 719–728.
- Gannon, M.C.; Nuttall, F.Q.; Lane, J.T.; Fang, S.; Gupta, V.; Sandhofer, C.R. Effect of 24 Hours of Starvation on Plasma Glucose and Insulin Concentrations in Subjects with Untreated Non—Insulin-Dependent Diabetes Mellitus. Metabolism 1996, 45, 492–497.
- Owen, O.E.; Felig, P.; Morgan, A.P.; Wahren, J.; Cahill, G.F. Liver and Kidney Metabolism during Prolonged Starvation. J. Clin. Investig. 1969, 48, 574–583.
- Cahill, G.F., Jr. Fuel Metabolism in Starvation. Annu. Rev. Nutr. 2006, 26, 1–22.
- Minich, T.; Yokota, S.; Dringen, R. Cytosolic and Mitochondrial Isoforms of NADP+-Dependent Isocitrate Dehydrogenases Are Expressed in Cultured Rat Neurons, Astrocytes, Oligodendrocytes and Microglial Cells. J. Neurochem. 2003, 86, 605–614.
- Kurz, G.M.; Wiesinger, H.; Hamprecht, B. Purification of Cytosolic Malic Enzyme from Bovine Brain, Generation of Monoclonal Antibodies, and Immunocytochemical Localization of the Enzyme in Glial Cells of Neural Primary Cultures. J. Neurochem. 1993, 60, 1467–1474.
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A.G. Cerebrospinal and Blood Levels of Amino Acids as Potential Biomarkers for Parkinson’s Disease: Review and Meta-analysis. Eur. J. Neurol. 2020, 27, 2336–2347.
- Verma, D.K.; Seo, B.A.; Ghosh, A.; Ma, S.-X.; Hernandez-Quijada, K.; Andersen, J.K.; Ko, H.S.; Kim, Y.-H. Alpha-Synuclein Preformed Fibrils Induce Cellular Senescence in Parkinson’s Disease Models. Cells 2021, 10, 1694.
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940.
- Martini, H.; Passos, J.F. Cellular Senescence: All Roads Lead to Mitochondria. FEBS J. 2022; early view.
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced MtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxidants 2021, 10, 1917.
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA Fragments Exit Mitochondria via MPTP- and VDAC-Dependent Channels to Activate NLRP3 Inflammasome and Interferon Signaling. Immunity, 2022, in press.
- Salnikov, V.; Lukyánenko, Y.O.; Frederick, C.A.; Lederer, W.J.; Lukyánenko, V. Probing the Outer Mitochondrial Membrane in Cardiac Mitochondria with Nanoparticles. Biophys. J. 2007, 92, 1058–1071.
- Rauckhorst, A.J.; Pfeiffer, D.R.; Broekemeier, K.M. The IPLA2γ Is Identified as the Membrane Potential Sensitive Phospholipase in Liver Mitochondria. FEBS Lett. 2015, 589, 2367–2371.
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A. Mitochondrial Permeability Transition Triggers the Release of MtDNA Fragments. CMLS Cell. Mol. Life Sci. 2004, 61, 3100–3103.
- García, N.; García, J.J.; Correa, F.; Chávez, E. The Permeability Transition Pore as a Pathway for the Release of Mitochondrial DNA. Life Sci. 2005, 76, 2873–2880.
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ Supplementation Reduces Neuroinflammation and Cell Senescence in a Transgenic Mouse Model of Alzheimer’s Disease via CGAS–STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118.
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular Mechanisms of Dietary Restriction Promoting Health and Longevity. Nat. Rev. Mol. Cell Biol. 2022, 23, 56–73.
- Mitchell, S.J.; Bernier, M.; Mattison, J.A.; Aon, M.A.; Kaiser, T.A.; Anson, R.M.; Ikeno, Y.; Anderson, R.M.; Ingram, D.K.; de Cabo, R. Daily Fasting Improves Health and Survival in Male Mice Independent of Diet Composition and Calories. Cell Metab. 2019, 29, 221–228.e3.
- Shimokawa, I.; Komatsu, T.; Hayashi, N.; Kim, S.-E.; Kawata, T.; Park, S.; Hayashi, H.; Yamaza, H.; Chiba, T.; Mori, R. The Life-Extending Effect of Dietary Restriction Requires Foxo3 in Mice. Aging Cell 2015, 14, 707–709.
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-Beta-Hydroxybutyrate Extends Lifespan in C. elegans. Aging 2014, 6, 621–644.
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent Metabolic Switching, Neuroplasticity and Brain Health. Nat. Rev. Neurosci. 2018, 19, 81–94.
- Hvingelby, V.S.; Glud, A.N.; Sørensen, J.C.H.; Tai, Y.; Andersen, A.S.M.; Johnsen, E.; Moro, E.; Pavese, N. Interventions to Improve Gait in Parkinson’s Disease: A Systematic Review of Randomized Controlled Trials and Network Meta-Analysis. J. Neurol. 2022, 269, 4068–4079.
- Uhrbrand, A.; Stenager, E.; Pedersen, M.S.; Dalgas, U. Parkinson’s Disease and Intensive Exercise Therapy—A Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Neurol. Sci. 2015, 353, 9–19.
- Stillman, C.M.; Cohen, J.; Lehman, M.E.; Erickson, K.I. Mediators of Physical Activity on Neurocognitive Function: A Review at Multiple Levels of Analysis. Front. Hum. Neurosci. 2016, 10, 626.
- Di Benedetto, S.; Müller, L.; Wenger, E.; Düzel, S.; Pawelec, G. Contribution of Neuroinflammation and Immunity to Brain Aging and the Mitigating Effects of Physical and Cognitive Interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128.
- Simeone, T.A.; Simeone, K.A.; Rho, J.M. Ketone Bodies as Anti-Seizure Agents. Neurochem. Res. 2017, 42, 2011–2018.
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 Year Multidomain Intervention of Diet, Exercise, Cognitive Training, and Vascular Risk Monitoring versus Control to Prevent Cognitive Decline in at-Risk Elderly People (FINGER): A Randomised Controlled Trial. Lancet 2015, 385, 2255–2263.
- Kwon, J.H.; Moon, K.M.; Min, K.-W. Exercise-Induced Myokines Can Explain the Importance of Physical Activity in the Elderly: An Overview. Healthcare 2020, 8, 378.
- Hao, Z.; Zhang, X.; Chen, P. Effects of Ten Different Exercise Interventions on Motor Function in Parkinson’s Disease Patients—A Network Meta-Analysis of Randomized Controlled Trials. Brain Sci. 2022, 12, 698.
- Gibala, M.J.; Little, J.P.; MacDonald, M.J.; Hawley, J.A. Physiological Adaptations to Low-Volume, High-Intensity Interval Training in Health and Disease. J. Physiol. 2012, 590, 1077–1084.
- Wahl, P.; Bloch, W.; Proschinger, S. The Molecular Signature of High-Intensity Training in the Human Body. Int. J. Sports Med. 2022, 43, 195–205.
- Ma, J.K.; Scribbans, T.D.; Edgett, B.A.; Boyd, J.C.; Simpson, C.A.; Little, J.P.; Gurd, B.J. Extremely Low-Volume, High-Intensity Interval Training Improves Exercise Capacity and Increases Mitochondrial Protein Content in Human Skeletal Muscle. Open J. Mol. Integr. Physiol. 2013, 3, 202–210.
- Sato, S.; Dyar, K.A.; Treebak, J.T.; Jepsen, S.L.; Ehrlich, A.M.; Ashcroft, S.P.; Trost, K.; Kunzke, T.; Prade, V.M.; Small, L.; et al. Atlas of Exercise Metabolism Reveals Time-Dependent Signatures of Metabolic Homeostasis. Cell Metab. 2022, 34, 329–345.e8.
- Williamson, D.H.; Lund, P.; Krebs, H.A. The Redox State of Free Nicotinamide-Adenine Dinucleotide in the Cytoplasm and Mitochondria of Rat Liver. Biochem. J. 1967, 103, 514–527.
- Goodman, R.P.; Markhard, A.L.; Shah, H.; Sharma, R.; Skinner, O.S.; Clish, C.B.; Deik, A.; Patgiri, A.; Hsu, Y.-H.; Masia, R.; et al. Hepatic NADH Reductive Stress Underlies Common Variation in Metabolic Traits. Nature 2020, 583, 122–126.
- Naghizadeh, F. Oxidation of Alpha-Hydroxybutyrate by Human Serum. Clin. Chim. Acta 1977, 81, 277–282.
- Perrin, L.; Loizides-Mangold, U.; Chanon, S.; Gobet, C.; Hulo, N.; Isenegger, L.; Weger, B.D.; Migliavacca, E.; Charpagne, A.; Betts, J.A.; et al. Transcriptomic Analyses Reveal Rhythmic and CLOCK-Driven Pathways in Human Skeletal Muscle. eLife 2018, 7, e34114.
- Schug, T.T.; Li, X. Sirtuin 1 in Lipid Metabolism and Obesity. Ann. Med. 2011, 43, 198–211.
- Luna, A.; McFadden, G.B.; Aladjem, M.I.; Kohn, K.W. Predicted Role of NAD Utilization in the Control of Circadian Rhythms during DNA Damage Response. PLoS Comput. Biol. 2015, 11, e1004144.
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.-K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian Clock Feedback Cycle Through NAMPT-Mediated NAD+ Biosynthesis. Science 2009, 324, 651–654.
- van Moorsel, D.; Hansen, J.; Havekes, B.; Scheer, F.A.J.L.; Jörgensen, J.A.; Hoeks, J.; Schrauwen-Hinderling, V.B.; Duez, H.; Lefebvre, P.; Schaper, N.C.; et al. Demonstration of a Day-Night Rhythm in Human Skeletal Muscle Oxidative Capacity. Mol. Metab. 2016, 5, 635–645.
- Qiu, Z.; Ming, H.; Lei, S.; Zhou, B.; Zhao, B.; Yu, Y.; Xue, R.; Xia, Z. Roles of HDAC3-Orchestrated Circadian Clock Gene Oscillations in Diabetic Rats Following Myocardial Ischaemia/Reperfusion Injury. Cell Death Dis. 2021, 12, 43.
- Parekh, P.K.; Ozburn, A.R.; McClung, C.A. Circadian Clock Genes: Effects on Dopamine, Reward and Addiction. Alcohol 2015, 49, 341–349.
- Breen, D.P.; Vuono, R.; Nawarathna, U.; Fisher, K.; Shneerson, J.M.; Reddy, A.B.; Barker, R.A. Sleep and Circadian Rhythm Regulation in Early Parkinson Disease. JAMA Neurol. 2014, 71, 589–595.
- Cai, Y.; Liu, S.; Sothern, R.B.; Xu, S.; Chan, P. Expression of Clock Genes Per1 and Bmal1 in Total Leukocytes in Health and Parkinson’s Disease: Clock Genes in PD. Eur. J. Neurol. 2010, 17, 550–554.
- Li, S.-Y.; Wang, Y.-L.; Liu, W.-W.; Lyu, D.-J.; Wang, F.; Mao, C.-J.; Yang, Y.-P.; Hu, L.-F.; Liu, C.-F. Long-Term Levodopa Treatment Accelerates the Circadian Rhythm Dysfunction in a 6-Hydroxydopamine Rat Model of Parkinson’s Disease. Chin. Med. J. 2017, 130, 1085–1092.
- Li, H.; Song, S.; Wang, Y.; Huang, C.; Zhang, F.; Liu, J.; Hong, J.-S. Low-Grade Inflammation Aggravates Rotenone Neurotoxicity and Disrupts Circadian Clock Gene Expression in Rats. Neurotox. Res. 2019, 35, 421–431.
- Liu, W.; Wei, S.; Huang, G.; Liu, L.; Gu, C.; Shen, Y.; Wang, X.; Xia, S.; Xie, A.; Hu, L.; et al. BMAL1 Regulation of Microglia-mediated Neuroinflammation in MPTP-induced Parkinson’s Disease Mouse Model. FASEB J. 2020, 34, 6570–6581.
- Kim, J.; Jang, S.; Choi, M.; Chung, S.; Choe, Y.; Choe, H.K.; Son, G.H.; Rhee, K.; Kim, K. Abrogation of the Circadian Nuclear Receptor REV-ERBα Exacerbates 6-Hydroxydopamine-Induced Dopaminergic Neurodegeneration. Mol. Cells 2018, 41, 742–752.
- de Goede, P.; Wefers, J.; Brombacher, E.C.; Schrauwen, P.; Kalsbeek, A. Circadian Rhythms in Mitochondrial Respiration. J. Mol. Endocrinol. 2018, 60, R115–R130.
- Jacobi, D.; Liu, S.; Burkewitz, K.; Kory, N.; Knudsen, N.H.; Alexander, R.K.; Unluturk, U.; Li, X.; Kong, X.; Hyde, A.; et al. Hepatic Bmal1 Regulates Rhythmic Mitochondrial Dynamics and Promotes Metabolic Fitness. Cell Metab. 2015, 22, 709–720.
- Schmitt, K.; Grimm, A.; Dallmann, R.; Oettinghaus, B.; Restelli, L.M.; Witzig, M.; Ishihara, N.; Mihara, K.; Ripperger, J.A.; Albrecht, U.; et al. Circadian Control of DRP1 Activity Regulates Mitochondrial Dynamics and Bioenergetics. Cell Metab. 2018, 27, 657–666.e5.
- Rabinovich-Nikitin, I.; Rasouli, M.; Reitz, C.J.; Posen, I.; Margulets, V.; Dhingra, R.; Khatua, T.N.; Thliveris, J.A.; Martino, T.A.; Kirshenbaum, L.A. Mitochondrial Autophagy and Cell Survival Is Regulated by the Circadian Clock Gene in Cardiac Myocytes during Ischemic Stress. Autophagy 2021, 17, 3794–3812.
- Galmozzi, A.; Mitro, N.; Ferrari, A.; Gers, E.; Gilardi, F.; Godio, C.; Cermenati, G.; Gualerzi, A.; Donetti, E.; Rotili, D.; et al. Inhibition of Class I Histone Deacetylases Unveils a Mitochondrial Signature and Enhances Oxidative Metabolism in Skeletal Muscle and Adipose Tissue. Diabetes 2013, 62, 732–742.
- Shen, R.-S.; Abell, C.W.; Gessner, W.; Brossi, A. Serotonergic Conversion of MPTP and Dopaminergic Accumulation of MPP+. FEBS Lett. 1985, 189, 225–230.
- Ramsay, R.R.; Dadgar, J.; Trevor, A.; Singer, T.P. Energy-Driven Uptake of N-Methyl-4-Phenylpyridine by Brain Mitochondria Mediates the Neurotoxicity of MPTP. Life Sci. 1986, 39, 581–588.
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100.
- Cassarino, D.S.; Parks, J.K.; Parker, W.D.; Bennett, J.P. The Parkinsonian Neurotoxin MPP+ Opens the Mitochondrial Permeability Transition Pore and Releases Cytochrome c in Isolated Mitochondria via an Oxidative Mechanism. Biochim. Biophys. Acta Mol. Basis Dis. 1999, 1453, 49–62.
- Klaidman, L.K.; Adams, J.D.; Leung, A.C.; Sam Kim, S.; Cadenas, E. Redox Cycling of MPP+: Evidence for a New Mechanism Involving Hydride Transfer with Xanthine Oxidase, Aldehyde Dehydrogenase, and Lipoamide Dehydrogenase. Free. Radic. Biol. Med. 1993, 15, 169–179.
- Adams, J.D., Jr.; Klaidman, L.K.; Leung, A.C. MPP+ and MPDP+ Induced Oxygen Radical Formation with Mitochondrial Enzymes. Free Radic. Biol. Med. 1993, 15, 181–186.
- Veech, R.L.; Todd King, M.; Pawlosky, R.; Kashiwaya, Y.; Bradshaw, P.C.; Curtis, W. The “Great” Controlling Nucleotide Coenzymes. IUBMB Life 2019, 71, 565–579.
- Makino, K.; Hagiwara, T.; Murakami, A. A Mini Review: Fundamental Aspects of Spin Trapping with DMPO. Int. J. Radiat. Appl. Instrum. Part C Radiat. Phys. Chem. 1991, 37, 657–665.
- Hoehne, M.N.; Jacobs, L.J.H.C.; Lapacz, K.J.; Calabrese, G.; Murschall, L.M.; Marker, T.; Kaul, H.; Trifunovic, A.; Morgan, B.; Fricker, M.; et al. Spatial and Temporal Control of Mitochondrial H2O2 Release in Intact Human Cells. EMBO J. 2022, 41, e109169.
- Xiao, B.; Deng, X.; Lim, G.G.Y.; Xie, S.; Zhou, Z.D.; Lim, K.-L.; Tan, E.-K. Superoxide Drives Progression of Parkin/PINK1-Dependent Mitophagy Following Translocation of Parkin to Mitochondria. Cell Death Dis. 2017, 8, e3097.
- Zhang, J.; Wang, B.; Wang, H.; He, H.; Wu, Q.; Qin, X.; Yang, X.; Chen, L.; Xu, G.; Yuan, Z.; et al. Disruption of the Superoxide Anions-Mitophagy Regulation Axis Mediates Copper Oxide Nanoparticles-Induced Vascular Endothelial Cell Death. Free Radic. Biol. Med. 2018, 129, 268–278.
- Aon, M.A.; Cortassa, S.; Marbán, E.; O’Rourke, B. Synchronized Whole Cell Oscillations in Mitochondrial Metabolism Triggered by a Local Release of Reactive Oxygen Species in Cardiac Myocytes. J. Biol. Chem. 2003, 278, 44735–44744.
- Marchissio, M.J.; Francés, D.E.A.; Carnovale, C.E.; Marinelli, R.A. Mitochondrial Aquaporin-8 Knockdown in Human Hepatoma HepG2 Cells Causes ROS-Induced Mitochondrial Depolarization and Loss of Viability. Toxicol. Appl. Pharmacol. 2012, 264, 246–254.
- Almasalmeh, A.; Krenc, D.; Wu, B.; Beitz, E. Structural Determinants of the Hydrogen Peroxide Permeability of Aquaporins. FEBS J. 2014, 281, 647–656.
- Wang, Y.; Yen, F.S.; Zhu, X.G.; Timson, R.C.; Weber, R.; Xing, C.; Liu, Y.; Allwein, B.; Luo, H.; Yeh, H.-W.; et al. SLC25A39 Is Necessary for Mitochondrial Glutathione Import in Mammalian Cells. Nature 2021, 599, 136–140.
- Gialluisi, A.; Reccia, M.G.; Modugno, N.; Nutile, T.; Lombardi, A.; Di Giovannantonio, L.G.; Pietracupa, S.; Ruggiero, D.; Scala, S.; Gambardella, S.; et al. Identification of Sixteen Novel Candidate Genes for Late Onset Parkinson’s Disease. Mol. Neurodegener. 2021, 16, 35.
- Francisco, A.; Engel, D.F.; Figueira, T.R.; Rogério, F.; de Bem, A.F.; Castilho, R.F. Mitochondrial NAD(P)+ Transhydrogenase Is Unevenly Distributed in Different Brain Regions, and Its Loss Causes Depressive-like Behavior and Motor Dysfunction in Mice. Neuroscience 2020, 440, 210–229.
- Lopes da Fonseca, T.; Villar-Piqué, A.; Outeiro, T.F. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 2015, 5, 435–471.
- Peth, A.; Nathan, J.A.; Goldberg, A.L. The ATP Costs and Time Required to Degrade Ubiquitinated Proteins by the 26 S Proteasome. J. Biol. Chem. 2013, 288, 29215–29222.
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-Dependent Steps in the Binding of Ubiquitin Conjugates to the 26S Proteasome That Commit to Degradation. Mol. Cell 2010, 40, 671–681.
- Singh, R.; Cuervo, A.M. Autophagy in the Cellular Energetic Balance. Cell Metab. 2011, 13, 495–504.
- Benderdour, M.; Charron, G.; deBlois, D.; Comte, B.; Rosiers, C.D. Cardiac Mitochondrial NADP+-Isocitrate Dehydrogenase Is Inactivated through 4-Hydroxynonenal Adduct Formation: An Event that Precedes Hypertrophy Development. J. Biol. Chem. 2003, 278, 45154–45159.
- Perez, M.A.; Magtanong, L.; Dixon, S.J.; Watts, J.L. Dietary Lipids Induce Ferroptosis in Caenorhabditis Elegans and Human Cancer Cells. Dev. Cell 2020, 54, 447–454.e4.
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis Driven by Radical Oxidation of N-6 Polyunsaturated Fatty Acids Mediates Acetaminophen-Induced Acute Liver Failure. Cell Death Dis. 2020, 11, 144.
- Tripathi, A.; Fanning, S.; Dettmer, U. Lipotoxicity Downstream of α-Synuclein Imbalance: A Relevant Pathomechanism in Synucleinopathies? Biomolecules 2021, 12, 40.
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c Acts as a Cardiolipin Oxygenase Required for Release of Proapoptotic Factors. Nat. Chem. Biol. 2005, 1, 223–232.
- Jenkins, C.M.; Yang, K.; Liu, G.; Moon, S.H.; Dilthey, B.G.; Gross, R.W. Cytochrome c Is an Oxidative Stress–Activated Plasmalogenase That Cleaves Plasmenylcholine and Plasmenylethanolamine at the Sn-1 Vinyl Ether Linkage. J. Biol. Chem. 2018, 293, 8693–8709.
- Hsu, Y.-H.; Dumlao, D.S.; Cao, J.; Dennis, E.A. Assessing Phospholipase A2 Activity toward Cardiolipin by Mass Spectrometry. PLoS ONE 2013, 8, e59267.
- Mancuso, D.J.; Kotzbauer, P.; Wozniak, D.F.; Sims, H.F.; Jenkins, C.M.; Guan, S.; Han, X.; Yang, K.; Sun, G.; Malik, I.; et al. Genetic Ablation of Calcium-Independent Phospholipase A2γ Leads to Alterations in Hippocampal Cardiolipin Content and Molecular Species Distribution, Mitochondrial Degeneration, Autophagy, and Cognitive Dysfunction. J. Biol. Chem. 2009, 284, 35632–35644.
- Mahajan, M.; Bharambe, N.; Shang, Y.; Lu, B.; Mandal, A.; Madan Mohan, P.; Wang, R.; Boatz, J.C.; Manuel Martinez Galvez, J.; Shnyrova, A.V.; et al. NMR Identification of a Conserved Drp1 Cardiolipin-Binding Motif Essential for Stress-Induced Mitochondrial Fission. Proc. Natl. Acad. Sci. USA 2021, 118, e2023079118.
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination Signal for Mitophagy in Neuronal Cells. Nat. Cell Biol. 2013, 15, 1197–1205.
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial Cardiolipin Is Required for Nlrp3 Inflammasome Activation. Immunity 2013, 39, 311–323.
- Brekk, O.R.; Honey, J.R.; Lee, S.; Hallett, P.J.; Isacson, O. Cell Type-Specific Lipid Storage Changes in Parkinson’s Disease Patient Brains Are Recapitulated by Experimental Glycolipid Disturbance. Proc. Natl. Acad. Sci. USA 2020, 117, 27646–27654.
- Ronchi, J.A.; Figueira, T.R.; Ravagnani, F.G.; Oliveira, H.C.F.; Vercesi, A.E.; Castilho, R.F. A Spontaneous Mutation in the Nicotinamide Nucleotide Transhydrogenase Gene of C57BL/6J Mice Results in Mitochondrial Redox Abnormalities. Free. Radic. Biol. Med. 2013, 63, 446–456.
- Zhu, J.; Schwörer, S.; Berisa, M.; Kyung, Y.J.; Ryu, K.W.; Yi, J.; Jiang, X.; Cross, J.R.; Thompson, C.B. Mitochondrial NADP(H) Generation Is Essential for Proline Biosynthesis. Science 2021, 372, 968–972.
- Abraham, M.A.; Lam, T.K.T. Glucagon Action in the Brain. Diabetologia 2016, 59, 1367–1371.
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavareá, J.M. The Identification of ATP-Citrate Lyase as a Protein Kinase B (Akt) Substrate in Primary Adipocytes. J. Biol. Chem. 2002, 277, 33895–33900.
- McReynolds, M.R.; Chellappa, K.; Chiles, E.; Jankowski, C.; Shen, Y.; Chen, L.; Descamps, H.C.; Mukherjee, S.; Bhat, Y.R.; Lingala, S.R.; et al. NAD+ Flux Is Maintained in Aged Mice despite Lower Tissue Concentrations. Cell Syst. 2021, 12, 1160–1172.e4.
- Veech, R.L.; Eggleston, L.V.; Krebs, H.A. The Redox State of Free Nicotinamide–Adenine Dinucleotide Phosphate in the Cytoplasm of Rat Liver. Biochem. J. 1969, 115, 609–619.
- Merrill, D.K.; Guynn, R.W. The Calculation of the Cytoplasmic Free / Ratio in Brain: Effect of Electroconvulsive Seizure. Brain Res. 1981, 221, 307–318.
- Subrahmanian, N.; LaVoie, M.J. Is There a Special Relationship between Complex I Activity and Nigral Neuronal Loss in Parkinson’s Disease? A Critical Reappraisal. Brain Res. 2021, 1767, 147434.
- Borst, P. The Malate–Aspartate Shuttle (Borst Cycle): How It Started and Developed into a Major Metabolic Pathway. IUBMB Life 2020, 72, 2241–2259.