Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Francisco Drusso Martinez Garcia | -- | 3094 | 2022-08-29 21:25:34 | | | |
| 2 | Vivi Li | + 197 word(s) | 3291 | 2022-08-30 04:51:55 | | | | |
| 3 | Vivi Li | Meta information modification | 3291 | 2022-08-31 08:29:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Martinez-Garcia, F.D.; Fischer, T.; Hayn, A.; Mierke, C.T.; Burgess, J.K.; Harmsen, M.C. Characterization of Hydrogel Microarchitecture for Cellular Applications. Encyclopedia. Available online: https://encyclopedia.pub/entry/26641 (accessed on 09 August 2026).
Martinez-Garcia FD, Fischer T, Hayn A, Mierke CT, Burgess JK, Harmsen MC. Characterization of Hydrogel Microarchitecture for Cellular Applications. Encyclopedia. Available at: https://encyclopedia.pub/entry/26641. Accessed August 09, 2026.
Martinez-Garcia, Francisco Drusso, Tony Fischer, Alexander Hayn, Claudia Tanja Mierke, Janette Kay Burgess, Martin Conrad Harmsen. "Characterization of Hydrogel Microarchitecture for Cellular Applications" Encyclopedia, https://encyclopedia.pub/entry/26641 (accessed August 09, 2026).
Martinez-Garcia, F.D., Fischer, T., Hayn, A., Mierke, C.T., Burgess, J.K., & Harmsen, M.C. (2022, August 29). Characterization of Hydrogel Microarchitecture for Cellular Applications. In Encyclopedia. https://encyclopedia.pub/entry/26641
Martinez-Garcia, Francisco Drusso, et al. "Characterization of Hydrogel Microarchitecture for Cellular Applications." Encyclopedia. Web. 29 August, 2022.
Copy Citation
The extracellular matrix (ECM) is a three-dimensional, acellular scaffold of living tissues. Incorporating the ECM into cell culture models is a goal of cell biology studies and requires biocompatible materials that can mimic the ECM. Among such materials are hydrogels: polymeric networks that derive most of their mass from water. With the tuning of their properties, these polymer networks can resemble living tissues. The microarchitectural properties of hydrogels, such as porosity, pore size, fiber length, and surface topology can determine cell plasticity. The adequate characterization of these parameters requires reliable and reproducible methods.
extracellular matrix
hydrogel architecture
topography
porosity
electron microscopy
laser microscopy
micro-computed tomography
atomic force microscopy
1. Introduction
The ECM is a three-dimensional (3D), acellular, heterogeneous network composed of fibrillar force-transducing collagens, interconnecting proteins such as fibronectin, matricellular proteins (e.g., periostin, fibulins, osteopontin), and the basement membrane proteins collagen type-IV and laminin [1]. Water retention is accomplished primarily by the highly negatively charged glycosaminoglycans (GAGs) or their higher order structures, i.e., GAGs bound to a protein core (proteoglycans), and to a lesser extent by collagens and similar proteins that also retain water [2]. The water concentration is highly tissue-specific, but it can range from 5% to 90% [3]. The ECM provides structural support and instruction to cells governed by its biophysical and biochemical cues.
Among the materials employed to mimic the ECM are hydrogels: highly porous, interconnected, hydrophilic, 3D polymeric networks that absorb and hold over 20% of their mass in water or other biological fluids [4][5]. When loaded with cells, hydrogels can provide biophysical conditions similar to those found in the native ECM [6][7]. For example, cell adhesion is not limited to a single plane, and there is no forced polarity as observed in vivo. Instead, in hydrogels, cell spreading and migration are modulated due to the variable stiffness and viscoelasticity of the material [8][9]. Hydrogels are formed via physical and chemical crosslinks and are commonly classified based on their polymer sources [4][10][11]. Fibrin [12], collagen [13], and decellularized organ-derived ECM [14][15][16][17][18] are examples of natural hydrogels and are regarded as biocompatible and bioactive [19]. These materials retain native cell-binding sites as well as protease-targeted degradation motifs, but due to their sources, batch-to-batch variations can influence the hydrogels’ tunability and overall mechanics and microarchitecture [10][19][20]. Synthetic hydrogels, such as polyacrylamide and polyethylene glycol (PEG), are regarded as more tunable than natural hydrogels, as their reconstitution conditions result in fewer batch-to-batch variations. Modifying the polymer backbones and molecular weights of synthetic hydrogels are common approaches to fine-tuning their properties. However, synthetic hydrogels lack inherent fundamental biological cues and require conjugation with cell-binding peptides (e.g., RGD, GFOGER, and IKVAV) to be biocompatible [10][21][22][23]. Semi-synthetic hydrogels, such as gelatin methacryloyl (GelMA) [24][25][26][27] or methacrylated hyaluronic acid (HAMA) [28][29], stem from the incorporation of crosslinking sites into the backbone of a natural polymer. These sites grant semi-synthetic hydrogels with a mechanical stability and tunability not commonly achieved in natural hydrogels [30][31][32]. Semi-synthetic hydrogels retain some biocompatible and bioactive features, being derivatives of natural polymers. For a more detailed description of the properties of individual hydrogel polymers, the reader is referred elsewhere [4][19][33]. The wide variety of polymers available for cell culture assays allow researchers to tailor hydrogel-based cell culture models to their research question(s). Within hydrogels, the microarchitecture is an inherent property known to influence the cell fate [34][35][36].
2. Electron-Based Techniques
2.1. Scanning Electron Microscopy
Scanning electron microscopy (SEM) is the most widely reported method for characterizing hydrogel microarchitectures [17][20][37][38][39][40][41][42][43][44][45][46]. This high-resolution imaging tool provides a detailed visualization of the hydrogel surface at the nanometer scale [47]. As its name suggests, SEM is an electron-based technique, where a high energy beam (aka “electron gun”) bombards a metal- or carbon-coated specimen with primary electrons, causing the emission of secondary and backscattered electrons. Secondary electrons highlight the morphology and topography of the specimen, while backscattered electrons provide contrast between areas with distinct chemical compositions (Figure 1). SEM imaging occurs under a high vacuum, as the presence of gas can attenuate the electron beams and stop them from scattering [48]. SEM microphotographs of hydrogels are employed to determine pore size, pore distribution, and porosity percentage, as well as fiber thickness and fiber orientation [17][37][38][41][42][43]. In cell-loaded hydrogels, the visualization of the cells is also possible [49][50][51][52][53]. The analytical capabilities of SEM include X-ray-based tools, such as energy dispersive X-Ray spectroscopy (EDX). EDX can detect elements such as C, S, O, N, Na, and others that are present in hydrogels [54][55][56][57] and within cells (e.g., P—a marker of DNA) (Figure 2A). EDX is particularly useful for the recognition of cells present within hydrogels of marked structural heterogeneity (Figure 2B). For more in-depth information on the use of EDX in biomedical research and diagnosis, the reader is referred elsewhere [58].
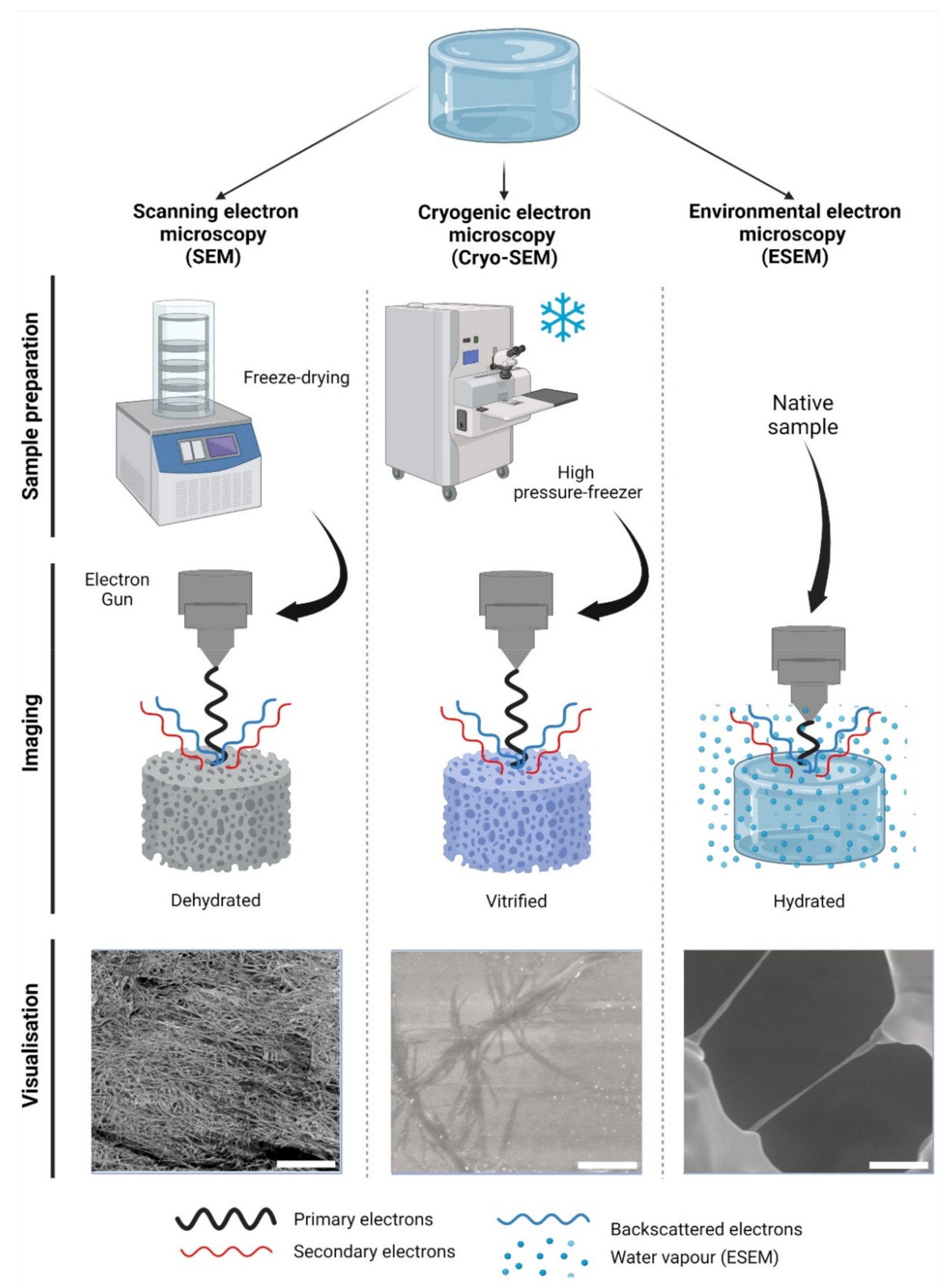
Figure 1. Electron-based imaging techniques for hydrogels. Standard scanning electron microscopy (SEM) relies on sample dehydration, including freeze-drying (shown) after fixation with aldehydes, followed by metal coating. Due to dehydration, the network appears condensed in visualization. In cryogenic SEM (Cryo-SEM), samples are vitrified using—among other methods—a high-pressure freezer (shown). Solid water ice is a source of imaging errors and is seen during imaging. In contrast, environmental SEM (ESEM) does not require a particular sample preparation, as it remains hydrated within a humidified chamber. In ESEM, the electron gun is closer to the specimen than in SEM or Cryo-SEM during imaging. As shown, single collagen fibers can be visualized; however, water condensation can cause imaging artefacts. The visualization of collagen type-I hydrogels (3.0 g/L) is shown at 12,000× magnification, 5 kV, and z = 9 mm. Scale bars represent 5 µm. Collagen type I hydrogels preparation, SEM, Cryo-SEM and ESEM detailed in Appendix A.

Figure 2. SEM-energy dispersive X-ray spectroscopy (EDX) elemental mapping. Images show human umbilical vein endothelial cells (HUVECs) on the surface of a gelatin methacryloyl (GelMA) hydrogel. (A) SEM allows for the visualization of cells (surrounded by a white dotted rectangle), while EDX identifies the elements present in both cells and hydrogels. These elements include non-metals such as phosphorus (P—a marker of DNA/nuclei), carbon (C), sulfur (S), and nitrogen (N). Transition metals used for coatings, such as gold (Au) and palladium (Pd), are also identified with EDX. Scale bars represent 10 µm and 25 µm. (B) In specimens with marked heterogeneity, EDX facilitates the identification of cells on the hydrogel surface (white dotted rectangle) that otherwise would not be distinguishable due to the condensation of the polymer network. Additional elements relevant to cell biology include non-metals, e.g., oxygen (O), and alkali metals, e.g., sodium (Na). Scale bars represent 50 µm and 100 µm. HUVEC’s culture conditions and GelMA properties detailed in Appendix A.
SEM-generated data indicate that higher polymer concentrations decrease pore sizes, but that cells are capable of modifying such porosity in hydrogels with degradation-sensitive sites [38]. In hydrogels that depend on functionalized groups to form crosslinks, SEM demonstrated that the degree of functionalization (DoF) has a greater influence on the pore density, pore size, and porosity percentage than the polymer concentration [59]. For example, high-DoF hydrogels have smaller pores than low-DoF hydrogels at similar polymer concentrations [30][59]. Moreover, both the polymer concentration and DoF have a direct influence on hydrogel swelling (i.e., water retention) and mechanics [38][43][59][60].
The limitations of SEM arise during the hydrogel preparation steps, as visualization requires a dry specimen. Thus, SEM is inherently biased, as desiccation will alter the native microarchitecture. Hydrogel desiccation is commonly achieved by passing a sample through a gradation of alcohol dehydration series [18][44][45][46][61][62] followed by freeze-drying [37][38][41][43][45][46] or critical point drying [17][61][62]. Thus, desiccation irreversibly alters the microarchitecture, leading to an imprecise hydrogel representation [46]. For example, collagen-HA hydrogels dried at −20 °C, −70 °C, and –196 °C showed variable (mean) pore sizes of 230, 90, and 40 μm, respectively [37]. Methods of applying fixatives such as glutaraldehyde [17][62] or combining with paraformaldehyde have been reported [20][61] for both cell-free and cell-loaded materials, but it is not clear to what extent the artefacts are prevented. Such artefacts can destroy finer features and leach out ions of interest [63]. Hydrogels are non-conductive, requiring irreversible carbon or metal coating (e.g., Au-Pd) [17][20][37][43][45][61] that could conceal finer surface details [63]. Despite these drawbacks, SEM data serve as a comparative measure when all hydrogels sustain the same systematic processing error. Moreover, SEM specimens can be preserved and visualized repeatedly, unlike samples imaged in other electron-based techniques, such as cryogenic SEM (Cryo-SEM) or environmental SEM (ESEM).
2.2. Cryogenic Scanning Electron Microscopy
Cryogenic scanning electron microscopy (Cryo-SEM) relies on a standard SEM with a field emission electron gun but employs a cryo-transfer system, where samples can be coated, fractured, and sublimated. In Cryo-SEM, samples must undergo vitrification: an ultra-rapid freezing method that prevents water crystal formation and generates a glass like-specimen [64]. The most widely reported method of vitrification in hydrogels is by plunge freezing either in liquid nitrogen, liquid ethane, liquid propane, or nitrogen slush at −137 °C [46][65][66][67]. Post-vitrification metal coating is not deemed essential but improves the imaging resolution [65]. Specimens can be fractured in order to visualize their inner-most microarchitecture and sublimated to remove the top layer of water, revealing the underlying microarchitecture [68]. The fast freezing step preserves biological structures with a higher fidelity than conventional SEM, rendering the Cryo-SEM imaging more factual in order to evaluate the hydrogel pore size [65], porosity [69], and fiber diameter (Figure 1) [68][69]. The presence of cells can also be detected [70][71]. As in SEM, most limitations of Cryo-SEM arise during the sample preparation stage. While, in principle, vitrification prevents ice crystallization, this process depends on a high cooling rate, which is difficult to achieve in specimens with a >10 µm thickness. The use of high-pressure freezing (Figure 1), which consists of a stream of liquid nitrogen at a rate of >2000 bar (~1.5 × 106 Torr) pressure can reportedly vitrify samples of a ≤500 µm thickness at −196 °C [64]. A poor cryo-fixation generates hexagonal ice crystals that displace the polymer network, causing structural damage [65]. Adding cryoprotectants improves the vitrification process, although the effective concentrations have been reported as cytotoxic upon prolonged exposure [67]. It is unclear whether cytotoxicity would cause any real alterations to cell-loaded specimens, as this step is performed immediately before freezing. Sublimation reportedly caused cracks on the surfaces of alginate hydrogels [65], and these are likely to occur in other polymer networks as well. Slow freezing rates are also reported, while sample dehydration and architecture distortion are common artefacts [65][66]. Compared to SEM, fewer studies report on the use of Cryo-SEM for hydrogels. For a detailed guide on hydrogel preparation for Cryo-SEM, the reader is directed elsewhere [65].
3. Photon-Based Techniques
3.1. Micro-Computed Tomography
Micro-computed tomography (µ-CT) is an X-ray-based scanning imaging tool that generates 2D trans-axial projections of a specimen [72][73]. While µ-CT is classified in this work as a photon-based technique, X-rays derive from the electron interactions within a high-energy electromagnetic beam [74]. In the µ-CT equipment, the sample is placed on a rotational stage and exposed to an X-ray source, and the passing light is captured by an X-ray detector. The passing X-rays can be attenuated (i.e., absorbed or scattered) by the sample thickness, density, and composition, providing phase contrast to the structures and components [75]. The µ-CT images can be reconstructed in 3D with an up to 1 μm voxel (3D pixel) resolution, making this a high-resolution technique. As an X-ray-based imaging method, µ-CT has historically been reported as a tool for reconstructing bone microarchitecture [76][77][78][79]. Studies on cell-loaded and cell-free hydrogels have used this technique to detect hydrogel mineralization both in vitro and in vivo [80][81][82][83]. The µ-CT data can be used to determine the pore size and fiber thickness in order to reconstruct the polymer network in 3D, demonstrating the pore interconnectivity [72][84]. Hydrogel degradation tests employing µ-CT in vitro demonstrate an increase in the pore sizes and porosity percentages [85][86][87][88]. While µ-CT is regarded as both non-destructive and non-invasive, exposure to high current and voltage levels will invariably dehydrate the sample [89]. High voltage levels are common when generating high-resolution images, but they are costly and result in a prolonged imaging time. To prevent structural damage during imaging, some reports recommend specimen fixation or the use of (freeze-)dried samples [80][87]. Thus, it is not uncommon to validate hydrogel µ-CT data using SEM data derived from dry specimens [85][89][90][91]. There are other limitations of µ-CT, as the hydrophilic nature of hydrogels provides a low-phase separation contrast (Figure 3) [83]. Radiopaque agents can be coupled with the polymer backbone or solubilized and left to soak with the hydrogel to improve said contrast [83]. Contrast agents allow researchers to discern between the hydrogel fibers and porosity and have been used to investigate hydrogel degradation in vivo and in vitro [92][93]. Reportedly, osmium tetroxide and uranyl acetate, or a combination of uranyl acetate and lead citrate, enabled a good contrast resolution and 3D reconstruction of collagen-I hydrogels [94]. The use of metal nanoparticles (e.g., Au) has also been reported to improve the hydrogel contrast [83] or the visualization of the spatial distribution of the cells within. Other limitations include vibrations in the µ-CT’s rotatory stage, which can cause motion artefacts by displacing the sample if it is not properly fixed—a common challenge with wet materials [89]. Information on the in-depth specifications and further limitations of µ-CT in materials research can be found elsewhere [75][89].
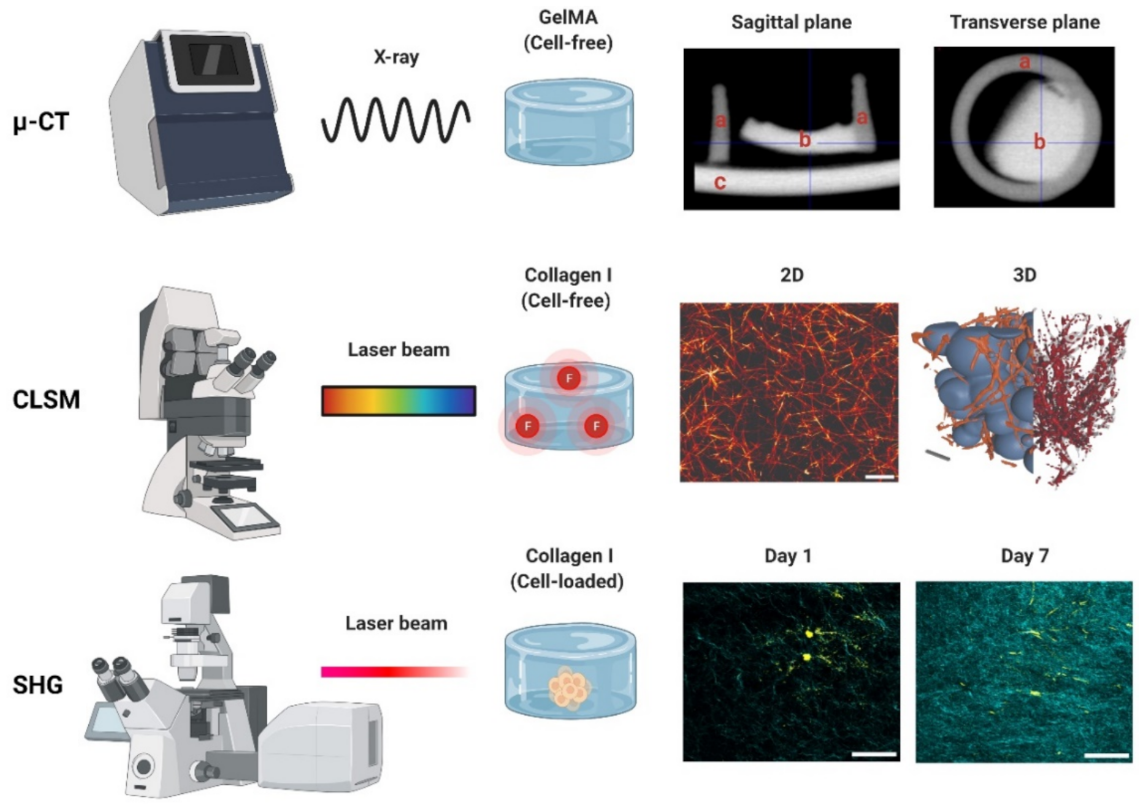
Figure 3. Photon-based imaging techniques. Micro-CT (µ-CT) employs X-rays, creating contrast based on the material properties (e.g., thickness, composition). A GelMA hydrogel scanned with µ-CT is shown in both the sagittal and transverse planes: (a) indicates the PCL container used for casting (2 mm diameter), (b) the GelMA hydrogel, and (c) the stage holder. Confocal laser scanning microscopy (CLSM) employs a laser to excite a fluorophore, which emits the fluorescent signal used for detection. Images of a collagen type-I hydrogel (3 g/L) are shown in 2D and 3D. The 3D image can be used to determine the interconnectivity of the polymer network (orange/red) using a bubble analysis (blue). Scale bars represent 5 µm (2D) and 10 µm (3D). Second harmonic generation (SHG). An example of a cell-loaded (fibroblast) collagen type-I hydrogel is shown on day 1 and day 7. The increase in the SHG signal indicates an increase in the collagen deposition during cell culture. SHG data derived from paraffin-embedded formalin-fixed hydrogels. Scale bars represent 50 µm. µ-CT, CLSM and SHG used to generate the data detailed in Appendix A.
3.2. Confocal Laser Scanning Microscopy
Confocal laser scanning microscopy (CLSM) is a photon-based fluorescent imaging technique. For visualization, CLSM requires that proteins and structures are stained or coupled with fluorophores: organic molecules that can emit light (λem) upon light excitation (λex) [95]. The CLSM optical resolution is determined by the wavelength of the laser. This wavelength is chosen according to the absorption spectra of the fluorophores, and multiple lasers can be used at the same time or in succession to capture multi-color fluorescent images [96]. Unlike conventional fluorescent microscopes that illuminate the entire specimen, CLSM uses point illumination, typically a laser beam, and a pinhole in front of the detector to eliminate most of the background blur, greatly increasing the optical resolution [97]. As only a single point of the specimen is illuminated, the samples must be scanned over a specified region to produce 2D or 3D fluorescent images.
CLSM is an invaluable tool used to visualize the structures of living cells within hydrogels [98]. For the assessment of the hydrogel microarchitecture, these can be fluorescently dyed [99], although certain polymers are naturally auto-fluorescent (e.g., chitosan), allowing dye-free imaging [100]. CLSM yields high-resolution 2D and 3D images (Figure 3), revealing the polymer network microarchitecture in great detail, crucial in the determination of the pore size and porosity of hydrogels in their swollen state (Figure 4) [99][101][102]. Overall, CLSM agrees with SEM in regards to the pore size decreasing with increasing polymer concentrations [99]. CLSM images can be segmented into a polymer phase and a fluid phase [103] (Figure 3, orange). During analysis, spheres can be fitted into the fluid phase (Figure 3, blue), and their diameter is measured to determine the pore diameter. The pore diameter, together with the median values of all the detected pore diameters, is defined as the pore size of a particular sample [103]. Applying the same principle to the polymer phase can determine the average thickness of individual fibers (Figure 4A). Additionally, CLSM images can be used to quantify local deviations in the microarchitecture due to nodes formed during the polymerization of collagen-I hydrogels. Moreover, data from fluorescently labelled collagen-I hydrogels showed that fiber diameter and fiber length are heavily influenced by pH, irrespective of the hydrogel concentration. In contrast, the pore size remained unaffected by pH [99].
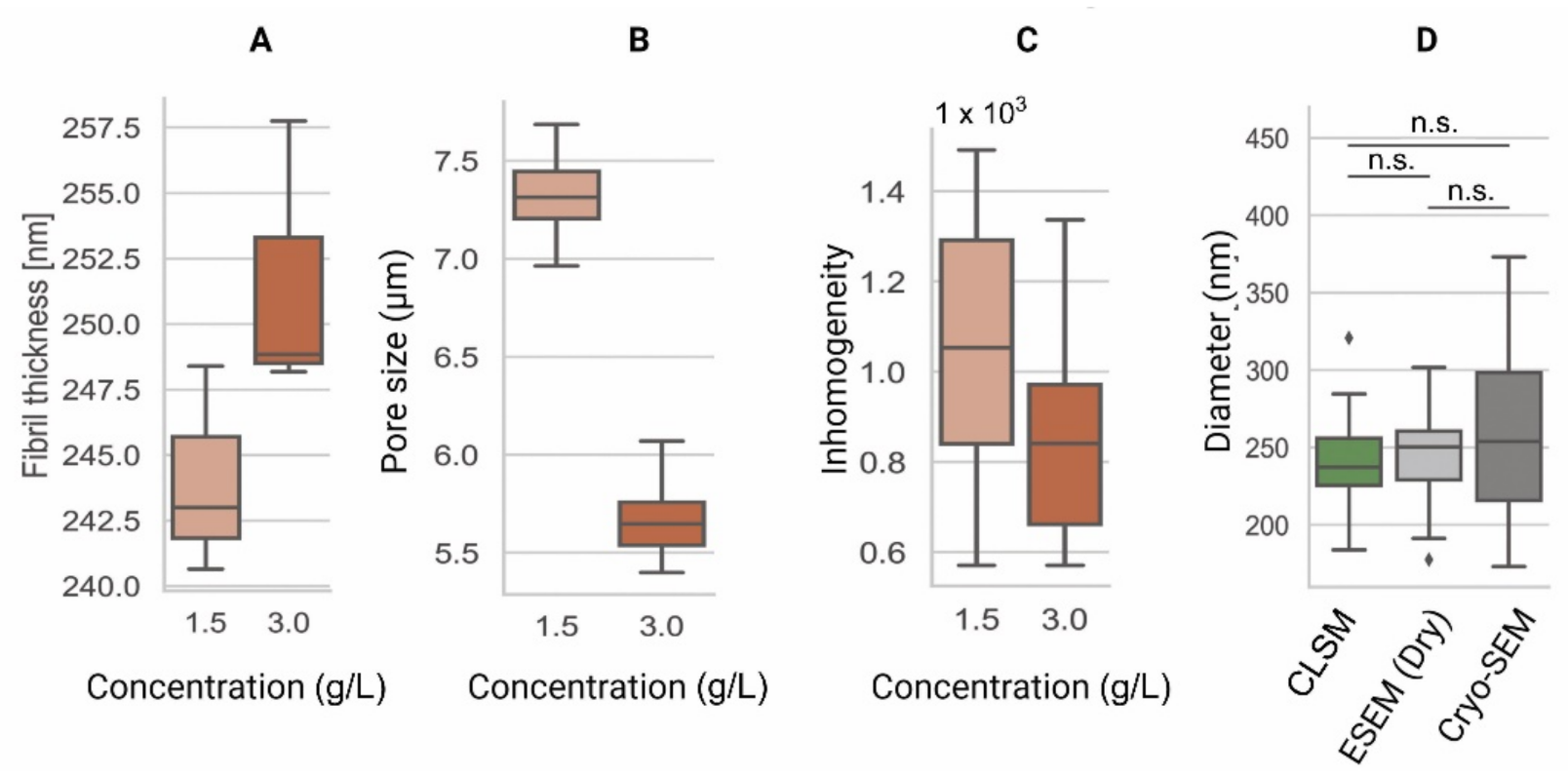
Figure 4. Comparison of the microarchitecture parameters among collagen type-I hydrogels of 1.5 g/L and 3.0 g/L. (A) Fiber thickness is an estimation of the 3D cross-sectional diameter of collagen fibers [104]. (B) Pore size represents the contiguous 3D space between collagen fibers [103]. (C) Inhomogeneity derived from the CLSM data is a measure of local and global variance in the pore size [101]. (D) Comparison of pore diameters among CLSM [103], ESEM, and Cryo-SEM images of collagen type-I hydrogels (3.0 g/L). These data indicate similar fiber diameter results among all techniques. Data shown in box-and-whisker plots indicating the median, first and third quartiles as boxes, variability as whiskers and outliers shown as ◆. n.s. = no significant differences.
Data from CLSM applied to cell-loaded hydrogels can be used to detect local cell-induced microenvironmental changes during migration through an inhomogeneity parameter [101]. This inhomogeneity parameter can explain differences in cell migration that cannot be explained using the pore size and mechanical properties alone. Therefore, elucidating these differences has led to significant insights into the role and adaptation of the microenvironment during cell migration [101].
CLSM has some limitations, the most important being the resolution limit due to optical diffraction limits. CLSM employs lasers with excitation wavelengths of several hundred nanometers, and the excited fluorophores commonly emit light at a higher wavelength. For example, collagen fibers in polymerized matrices possess a wide range of diameters, ranging from below 100 nm [105] to nearly 1 µm [99]. Using a λex 561 nm-laser and recording λem 580 nm, objects as small as 290 nm can be distinguished. Thus, this technique [103] must be considered an overestimation, as the fiber thickness can be below the optical diffraction limit. However, due to multiple post-processing steps, the analysis can be considered as quasi-pixelwise. Another important consideration is the efficacy of the fluorescent dyes used. Poor staining might result in a low signal-to-noise ratio or even the inhomogeneous staining of a hydrogel, resulting in limitations in the dynamic range. Thicker and, thus, brighter fibers may overlay smaller fibrils, or vice versa, with larger fibers losing detail when overexposed. Lastly, the choice of a spherical space estimator might not be suitable for a specific problem. For a robust estimate of the cell migration, spheres can be considered a suitable shape. However, other problems can make it necessary to choose other shapes, such as cubes. Fitting shapes into the hydrogel segmentation might be entirely superfluous, and polymer content calculations based on the precise segmentation may provide a better parameter.
Appendix A
Collagen-I hydrogels (3.0 g/L) shown in Figure 1, Figure 2, Figure 3, Figure 4 were prepared as described here [106]. For the CLSM imaging, the collagen-I hydrogels were polymerized on top of a functionalized glass-coverslip and coated with (3-Aminopropyl)trimethoxysilane (APTMS; Sigma Aldrich, St Louis, MO, USA). The GelMA hydrogels (5% w/v) shown in Figure 2 and Figure 3 are derived from the same batch reported here [63], with a 56.0 ± 0.9% DoF according to nuclear magnetic resonance.
The HUVECs shown in Figure 2 were cultured in RPMI 1640 (Lonza, Verviers, Belgium) supplemented with 20% FBS (Gibco, Paisley, UK), 1% Pen-Strep (Gibco), 2 mM L-Glut (BioWhittaker, Walkersville, MD, USA), 5 U/mL Heparin (Leo Pharma, Amsterdam, the Netherlands), and 50 µg/mL crude endothelial cell growth factor (Roche, Mannheim, Germany).
The data presented in this entry were obtained with the following instruments: SEM-Zeiss Supra 55 STEM (Carl Zeiss NTS GmbH, Oberkochen, Germany); Cryo-SEM and ESEM-Quanta 200 FEG (Philips-FEI, Hillsboro, OR, USA); µCT-Inveon PET/CT (Siemens Medical Solutions USA, Knoxville, TN, USA); CLSM-Zeiss LSM 780 (Carl Zeiss NTS GmbH, Oberkochen, Germany); SHG-SP5 multiphoton (Leica, Amsterdam, The Netherlands); and AFM-JPK CellHesion® R200 (Bruker Nano GmbH, Berlin, Germany) with Arrow-TL2-50 cantilevers (Nano-World®, Neuchâtel, Switzerland) and polystyrene beads glued to them. All data shown are original and were not published elsewhere.
References
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The Extracellular Matrix at a Glance. J. Cell Sci. 2010, 123, 4195–4200.
- Couchman, J.R.; Pataki, C.A. An Introduction to Proteoglycans and Their Localization. J. Histochem. Cytochem. 2012, 60, 885–897.
- Pethig, R.; Kell, D.B. The Passive Electrical Properties of Biological Systems: Their Significance in Physiology, Biophysics and Biotechnology. Phys. Med. Biol. 1987, 32, 933–970.
- Ahmed, E.M. Hydrogel: Preparation, Characterization, and Applications: A Review. J. Adv. Res. 2015, 6, 105–121.
- Pita-López, M.L.; Fletes-Vargas, G.; Espinosa-Andrews, H.; Rodríguez-Rodríguez, R. Physically Cross-Linked Chitosan-Based Hydrogels for Tissue Engineering Applications: A State-of-the-Art Review. Eur. Polym. J. 2021, 145, 110176.
- Lang, N.R.; Skodzek, K.; Hurst, S.; Mainka, A.; Steinwachs, J.; Schneider, J.; Aifantis, K.E.; Fabry, B. Biphasic Response of Cell Invasion to Matrix Stiffness in Three-Dimensional Biopolymer Networks. Acta Biomater. 2015, 13, 61–67.
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional Homeostasis and the Malignant Phenotype. Cancer Cell 2005, 8, 241–254.
- Li, Y.; Kilian, K.A. Bridging the Gap: From 2D Cell Culture to 3D Microengineered Extracellular Matrices. Adv. Healthc. Mater. 2015, 4, 2780–2796.
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as Extracellular Matrix Mimics for 3D Cell Culture. Biotechnol. Bioeng. 2009, 103, 655–663.
- Tsou, Y.H.; Khoneisser, J.; Huang, P.C.; Xu, X. Hydrogel as a Bioactive Material to Regulate Stem Cell Fate. Bioact. Mater. 2016, 1, 39–55.
- Ruedinger, F.; Lavrentieva, A.; Blume, C.; Pepelanova, I.; Scheper, T. Hydrogels for 3D Mammalian Cell Culture: A Starting Guide for Laboratory Practice. Appl. Microbiol. Biotechnol. 2015, 99, 623–636.
- Stanton, M.M.; Samitier, J.; Sánchez, S. Bioprinting of 3D Hydrogels. Lab A Chip 2015, 15, 3111–3115.
- Kim, H.-D.; Guo, T.W.; Wu, A.P.; Wells, A.; Gertler, F.B.; Lauffenburger, D.A. Epidermal Growth Factor–Induced Enhancement of Glioblastoma Cell Migration in 3D Arises from an Intrinsic Increase in Speed But an Extrinsic Matrix- and Proteolysis-Dependent Increase in Persistence. Mol. Biol. Cell 2008, 19, 4249–4259.
- De Hilster, R.H.J.; Sharma, P.K.; Jonker, M.R.; White, E.S.; Gercama, E.A.; Roobeek, M.; Timens, W.; Harmsen, M.C.; Hylkema, M.N.; Burgess, J.K. Human Lung Extracellular Matrix Hydrogels Resemble the Stiffness and Viscoelasticity of Native Lung Tissue. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2020, 318, L698–L704.
- Liguori, G.R.; Liguori, T.T.A.; de Moraes, S.R.; Sinkunas, V.; Terlizzi, V.; van Dongen, J.A.; Sharma, P.K.; Moreira, L.F.P.; Harmsen, M.C. Molecular and Biomechanical Clues From Cardiac Tissue Decellularized Extracellular Matrix Drive Stromal Cell Plasticity. Front. Bioeng. Biotechnol. 2020, 8, 520.
- Dongen, J.A.; Getova, V.; Brouwer, L.A.; Liguori, G.R.; Sharma, P.K.; Stevens, H.P.; Lei, B.; Harmsen, M.C. Adipose Tissue-derived Extracellular Matrix Hydrogels as a Release Platform for Secreted Paracrine Factors. J. Tissue Eng. Regen. Med. 2019, 13, 973–985.
- Freytes, D.O.; Martin, J.; Velankar, S.S.; Lee, A.S.; Badylak, S.F. Preparation and Rheological Characterization of a Gel Form of the Porcine Urinary Bladder Matrix. Biomaterials 2008, 29, 1630–1637.
- Sackett, S.D.; Tremmel, D.M.; Ma, F.; Feeney, A.K.; Maguire, R.M.; Brown, M.E.; Zhou, Y.; Li, X.; O’Brien, C.; Li, L.; et al. Extracellular Matrix Scaffold and Hydrogel Derived from Decellularized and Delipidized Human Pancreas. Sci. Rep. 2018, 8, 10452.
- Catoira, M.C.; Fusaro, L.; Di Francesco, D.; Ramella, M.; Boccafoschi, F. Overview of Natural Hydrogels for Regenerative Medicine Applications. J. Mater. Sci. Mater. Med. 2019, 30, 115.
- Martinez-Garcia, F.D.; de Hilster, R.H.J.; Sharma, P.K.; Borghuis, T.; Hylkema, M.N.; Burgess, J.K.; Harmsen, M.C. Architecture and Composition Dictate Viscoelastic Properties of Organ-Derived Extracellular Matrix Hydrogels. Polymers 2021, 13, 3113.
- Wang, Y.; Zhao, Q.; Zhang, H.; Yang, S.; Jia, X. A Novel Poly(Amido Amine)-Dendrimer-Based Hydrogel as a Mimic for the Extracellular Matrix. Adv. Mater. 2014, 26, 4163–4167.
- Collier, J.H.; Segura, T. Evolving the Use of Peptides as Components of Biomaterials. Biomaterials 2011, 32, 4198–4204.
- Ki, C.S.; Lin, T.-Y.; Korc, M.; Lin, C.-C. Thiol-Ene Hydrogels as Desmoplasia-Mimetic Matrices for Modeling Pancreatic Cancer Cell Growth, Invasion, and Drug Resistance. Biomaterials 2014, 35, 9668–9677.
- Krishnamoorthy, S.; Noorani, B.; Xu, C. Effects of Encapsulated Cells on the Physical–Mechanical Properties and Microstructure of Gelatin Methacrylate Hydrogels. Int. J. Mol. Sci. 2019, 20, 5061.
- Loessner, D.; Meinert, C.; Kaemmerer, E.; Martine, L.C.; Yue, K.; Levett, P.A.; Klein, T.J.; Melchels, F.P.W.; Khademhosseini, A.; Hutmacher, D.W. Functionalization, Preparation and Use of Cell-Laden Gelatin Methacryloyl-Based Hydrogels as Modular Tissue Culture Platforms. Nat. Protoc. 2016, 11, 727–746.
- Yoon, H.J.; Shin, S.R.; Cha, J.M.; Lee, S.H.; Kim, J.H.; Do, J.T.; Song, H.; Bae, H. Cold Water Fish Gelatin Methacryloyl Hydrogel for Tissue Engineering Application. PLoS ONE 2016, 11, 1–18.
- Li, X.; Zhang, J.; Kawazoe, N.; Chen, G. Fabrication of Highly Crosslinked Gelatin Hydrogel and Its Influence on Chondrocyte Proliferation and Phenotype. Polymers 2017, 9, 309.
- Kessler, L.; Gehrke, S.; Winnefeld, M.; Huber, B.; Hoch, E.; Walter, T.; Wyrwa, R.; Schnabelrauch, M.; Schmidt, M.; Kückelhaus, M.; et al. Methacrylated Gelatin/Hyaluronan-Based Hydrogels for Soft Tissue Engineering. J. Tissue Eng. 2017, 8, 204173141774415.
- Camci-Unal, G.; Cuttica, D.; Annabi, N.; Demarchi, D.; Khademhosseini, A. Synthesis and Characterization of Hybrid Hyaluronic Acid-Gelatin Hydrogels. Biomacromolecules 2013, 14, 1085–1092.
- Sun, M.; Sun, X.; Wang, Z.; Guo, S.; Yu, G.; Yang, H. Synthesis and Properties of Gelatin Methacryloyl (GelMA) Hydrogels and Their Recent Applications in Load-Bearing Tissue. Polymers 2018, 10, 1290.
- Yin, J.; Yan, M.; Wang, Y.; Fu, J.; Suo, H. 3D Bioprinting of Low-Concentration Cell-Laden Gelatin Methacrylate (GelMA) Bioinks with a Two-Step Cross-Linking Strategy. ACS Appl. Mater. Interfaces 2018, 10, 6849–6857.
- Pepelanova, I.; Kruppa, K.; Scheper, T.; Lavrentieva, A. Gelatin-Methacryloyl (GelMA) Hydrogels with Defined Degree of Functionalization as a Versatile Toolkit for 3D Cell Culture and Extrusion Bioprinting. Bioengineering 2018, 5, 55.
- Madduma-Bandarage, U.S.K.; Madihally, S.V. Synthetic Hydrogels: Synthesis, Novel Trends, and Applications. J. Appl. Polym. Sci. 2021, 138, 50376.
- Chaudhuri, O.; Gu, L.; Klumpers, D.; Darnell, M.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Lee, H.; Lippens, E.; Duda, G.N.; et al. Hydrogels with Tunable Stress Relaxation Regulate Stem Cell Fate and Activity. Nat. Mater. 2015, 15, 326.
- Chaudhuri, O.; Gu, L.; Darnell, M.; Klumpers, D.; Bencherif, S.A.; Weaver, J.C.; Huebsch, N.; Mooney, D.J. Substrate Stress Relaxation Regulates Cell Spreading. Nat. Commun. 2015, 6, 6365.
- Wisdom, K.M.; Adebowale, K.; Chang, J.; Lee, J.Y.; Nam, S.; Desai, R.; Rossen, N.S.; Rafat, M.; West, R.B.; Hodgson, L.; et al. Matrix Mechanical Plasticity Regulates Cancer Cell Migration through Confining Microenvironments. Nat. Commun. 2018, 9, 4144.
- Park, S.-N.; Park, J.-C.; Kim, H.O.; Song, M.J.; Suh, H. Characterization of Porous Collagen/Hyaluronic Acid Scaffold Modified by 1-Ethyl-3-(3-Dimethylaminopropyl)Carbodiimide Cross-Linking. Biomaterials 2002, 23, 1205–1212.
- Chen, Y.; Lin, R.; Qi, H.; Yang, Y.; Bae, H.; Melero-Martin, J.M.; Khademhosseini, A. Functional Human Vascular Network Generated in Photocrosslinkable Gelatin Methacrylate Hydrogels. Adv. Funct. Mater. 2012, 22, 2027–2039.
- Chavda, H.; Modhia, I.; Patel, R.; Patel, C. Preparation and Characterization of Superporous Hydrogel Based on Different Polymers. Int. J. Pharm. Investig. 2012, 2, 134.
- Van Vlierberghe, S.; Cnudde, V.; Dubruel, P.; Masschaele, B.; Cosijns, A.; de Paepe, I.; Jacobs, P.J.S.; van Hoorebeke, L.; Remon, J.P.; Schacht, E. Porous Gelatin Hydrogels: 1. Cryogenic Formation and Structure Analysis. Biomacromolecules 2007, 8, 331–337.
- Xiao, W.; He, J.; Nichol, J.W.; Wang, L.; Hutson, C.B.; Wang, B.; Du, Y.; Fan, H.; Khademhosseini, A. Synthesis and Characterization of Photocrosslinkable Gelatin and Silk Fibroin Interpenetrating Polymer Network Hydrogels. Acta Biomater. 2011, 7, 2384–2393.
- Choi, S.-W.; Xie, J.; Xia, Y. Chitosan-Based Inverse Opals: Three-Dimensional Scaffolds with Uniform Pore Structures for Cell Culture. Adv. Mater. 2009, 21, 2997–3001.
- Eke, G.; Mangir, N.; Hasirci, N.; MacNeil, S.; Hasirci, V. Development of a UV Crosslinked Biodegradable Hydrogel Containing Adipose Derived Stem Cells to Promote Vascularization for Skin Wounds and Tissue Engineering. Biomaterials 2017, 129, 188–198.
- Habib, A.; Sathish, V.; Mallik, S.; Khoda, B. 3D Printability of Alginate-Carboxymethyl Cellulose Hydrogel. Materials 2018, 11, 454.
- Jia, W.; Gungor-Ozkerim, P.S.; Zhang, Y.S.; Yue, K.; Zhu, K.; Liu, W.; Pi, Q.; Byambaa, B.; Dokmeci, M.R.; Shin, S.R.; et al. Direct 3D Bioprinting of Perfusable Vascular Constructs Using a Blend Bioink. Biomaterials 2016, 106, 58–68.
- Koch, M.; Włodarczyk-Biegun, M.K. Faithful Scanning Electron Microscopic (SEM) Visualization of 3D Printed Alginate-Based Scaffolds. Bioprinting 2020, 20, e00098.
- Doucet, F.J.; Lead, J.R.; Maguire, L.; Achterberg, E.P.; Millward, G.E. Visualisation of Natural Aquatic Colloids and Particles–A Comparison of Conventional High Vacuum and Environmental Scanning Electron Microscopy. J. Environ. Monitor. 2005, 7, 115.
- Donald, A.M. The Use of Environmental Scanning Electron Microscopy for Imaging Wet and Insulating Materials. Nat. Mater. 2003, 2, 511–516.
- Zheng, H.; Tian, W.; Yan, H.; Yue, L.; Zhang, Y.; Han, F.; Chen, X.; Li, Y. Rotary Culture Promotes the Proliferation of MCF-7 Cells Encapsulated in Three-Dimensional Collagen–Alginate Hydrogels via Activation of the ERK1/2-MAPK Pathway. Biomed. Mater. 2012, 7, 015003.
- Liang, R.; Yang, G.; Kim, K.E.; D’Amore, A.; Pickering, A.N.; Zhang, C.; Woo, S.L.-Y. Positive Effects of an Extracellular Matrix Hydrogel on Rat Anterior Cruciate Ligament Fibroblast Proliferation and Collagen MRNA Expression. J. Orthop. Transl. 2015, 3, 114–122.
- Eslami, M.; Vrana, N.E.; Zorlutuna, P.; Sant, S.; Jung, S.; Masoumi, N.; Khavari-Nejad, R.A.; Javadi, G.; Khademhosseini, A. Fiber-Reinforced Hydrogel Scaffolds for Heart Valve Tissue Engineering. J. Biomater. Appl. 2014, 29, 399–410.
- Zhong, X.; Ji, C.; Chan, A.K.L.; Kazarian, S.G.; Ruys, A.; Dehghani, F. Fabrication of Chitosan/Poly(ε-Caprolactone) Composite Hydrogels for Tissue Engineering Applications. J. Mater. Sci. Mater. Med. 2011, 22, 279–288.
- Patiño Vargas, M.I.; Martinez-Garcia, F.D.; Offens, F.; Becerra, N.Y.; Restrepo, L.M.; van der Mei, H.C.; Harmsen, M.C.; van Kooten, T.G.; Sharma, P.K. Viscoelastic Properties of Plasma-Agarose Hydrogels Dictate Favorable Fibroblast Responses for Skin Tissue Engineering Applications. Biomater. Adv. 2022, 139, 212967.
- Sattari, S.; Dadkhah Tehrani, A.; Adeli, M. PH-Responsive Hybrid Hydrogels as Antibacterial and Drug Delivery Systems. Polymers 2018, 10, 660.
- Vilela, P.B.; Dalalibera, A.; Becegato, V.A.; Paulino, A.T. Single-Component and Multi-Component Metal Abatement in Water Using a Hydrogel Based on Chitosan: Characterization, Isotherm, Kinetic, and Thermodynamic Results. Water Air Soil Pollut. 2020, 231, 507.
- Guven, M.N.; Seckin Altuncu, M.; Demir Duman, F.; Eren, T.N.; Yagci Acar, H.; Avci, D. Bisphosphonate-Functionalized Poly(β-Amino Ester) Network Polymers. J. Biomed. Mater. Res. 2017, 105, 1412–1421.
- Ha, J.H.; Lim, J.H.; Kim, J.W.; Cho, H.-Y.; Jo, S.G.; Lee, S.H.; Eom, J.Y.; Lee, J.M.; Chung, B.G. Conductive GelMA–Collagen–AgNW Blended Hydrogel for Smart Actuator. Polymers 2021, 13, 1217.
- Scimeca, M.; Bischetti, S.; Lamsira, H.K.; Bonfiglio, R.; Bonanno, E. Energy Dispersive X-Ray (EDX) Microanalysis: A Powerful Tool in Biomedical Research and Diagnosis. Eur. J. Histochem. 2018, 62, 2841.
- Nichol, J.W.; Koshy, S.T.; Bae, H.; Hwang, C.M.; Yamanlar, S.; Khademhosseini, A. Cell-Laden Microengineered Gelatin Methacrylate Hydrogels. Biomaterials 2010, 31, 5536–5544.
- Martinez-Garcia, F.D.; Valk, M.M.; Sharma, P.K.; Burgess, J.K.; Harmsen, M.C. Adipose Tissue-Derived Stromal Cells Alter the Mechanical Stability and Viscoelastic Properties of Gelatine Methacryloyl Hydrogels. IJMS 2021, 22, 10153.
- Laronda, M.M.; Rutz, A.L.; Xiao, S.; Whelan, K.A.; Duncan, F.E.; Roth, E.W.; Woodruff, T.K.; Shah, R.N. A Bioprosthetic Ovary Created Using 3D Printed Microporous Scaffolds Restores Ovarian Function in Sterilized Mice. Nat. Commun. 2017, 8, 15261.
- Chimenti, I.; Rizzitelli, G.; Gaetani, R.; Angelini, F.; Ionta, V.; Forte, E.; Frati, G.; Schussler, O.; Barbetta, A.; Messina, E.; et al. Human Cardiosphere-Seeded Gelatin and Collagen Scaffolds as Cardiogenic Engineered Bioconstructs. Biomaterials 2011, 32, 9271–9281.
- McKinlay, K.J.; Allison, F.J.; Scotchford, C.A.; Grant, D.M.; Oliver, J.M.; King, J.R.; Wood, J.V.; Brown, P.D. Comparison of Environmental Scanning Electron Microscopy with High Vacuum Scanning Electron Microscopy as Applied to the Assessment of Cell Morphology. J. Biomed. Mater. Res. 2004, 69, 359–366.
- Bokstad, M.; Medalia, O. Correlative Light Electron Microscopy as a Navigating Tool for Cryo-Electron Tomography Analysis. In Fluorescence Microscopy; Elsevier: Amsterdam, The Netherlands, 2014; pp. 121–131. ISBN 978-0-12-409513-7.
- Aston, R.; Sewell, K.; Klein, T.; Lawrie, G.; Grøndahl, L. Evaluation of the Impact of Freezing Preparation Techniques on the Characterisation of Alginate Hydrogels by Cryo-SEM. Eur. Polym. J. 2016, 82, 1–15.
- Kuleshova, L.L.; Gouk, S.S.; Hutmacher, D.W. Vitrification as a Prospect for Cryopreservation of Tissue-Engineered Constructs. Biomaterials 2007, 28, 1585–1596.
- Tavukcuoglu, S.; Al-Azawi, T.; Khaki, A.A.; Al-Hasani, S. Is Vitrification Standard Method of Cryopreservation. Middle East Fertil. Soc. J. 2012, 17, 152–156.
- Ivan’kova, E.M.; Dobrovolskaya, I.P.; Popryadukhin, P.V.; Kryukov, A.; Yudin, V.E.; Morganti, P. In-Situ Cryo-SEM Investigation of Porous Structure Formation of Chitosan Sponges. Polym. Test. 2016, 52, 41–45.
- Ji, C.; Annabi, N.; Khademhosseini, A.; Dehghani, F. Fabrication of Porous Chitosan Scaffolds for Soft Tissue Engineering Using Dense Gas CO2. Acta Biomater. 2011, 7, 1653–1664.
- Schnabel-Lubovsky, M.; Kossover, O.; Melino, S.; Nanni, F.; Talmon, Y.; Seliktar, D. Visualizing Cell-laden Fibrin-based Hydrogels Using Cryogenic Scanning Electron Microscopy and Confocal Microscopy. J. Tissue Eng. Regen. Med. 2019, 13, 587–598.
- Valot, L.; Maumus, M.; Brunel, L.; Martinez, J.; Amblard, M.; Noël, D.; Mehdi, A.; Subra, G. A Collagen-Mimetic Organic-Indorganic Hydrogel for Cartilage Engineering. Gels 2021, 7, 73.
- Van Vlierberghe, S.; Dubruel, P.; Lippens, E.; Masschaele, B.; van Hoorebeke, L.; Cornelissen, M.; Unger, R.; Kirkpatrick, C.J.; Schacht, E. Toward Modulating the Architecture of Hydrogel Scaffolds: Curtains versus Channels. J. Mater. Sci. Mater. Med. 2008, 19, 1459–1466.
- Boerckel, J.D.; Mason, D.E.; McDermott, A.M.; Alsberg, E. Microcomputed Tomography: Approaches and Applications in Bioengineering. Stem Cell Res. Ther. 2014, 5, 144.
- Morelhão, S.L. Fundamentals of X-Ray Physics. In Computer Simulation Tools for X-ray Analysis; Graduate Texts in Physics; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–57. ISBN 978-3-319-19553-7.
- Olăreț, E.; Stancu, I.-C.; Iovu, H.; Serafim, A. Computed Tomography as a Characterization Tool for Engineered Scaffolds with Biomedical Applications. Materials 2021, 14, 6763.
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Müller, R. Guidelines for Assessment of Bone Microstructure in Rodents Using Micro-Computed Tomography. J. Bone Miner. Res. 2010, 25, 1468–1486.
- Wu, Y.; Adeeb, S.; Doschak, M.R. Using Micro-CT Derived Bone Microarchitecture to Analyze Bone Stiffness €“ A Case Study on Osteoporosis Rat Bone. Front. Endocrinol. 2015, 6, 80.
- Hua, Y.; Bi, R.; Zhang, Y.; Xu, L.; Guo, J.; Li, Y. Different Bone Sites-Specific Response to Diabetes Rat Models: Bone Density, Histology and Microarchitecture. PLoS ONE 2018, 13, e0205503.
- Feldkamp, L.A.; Goldstein, S.A.; Parfitt, M.A.; Jesion, G.; Kleerekoper, M. The Direct Examination of Three-Dimensional Bone Architecture in Vitro by Computed Tomography. J. Bone Miner. Res. 2009, 4, 3–11.
- Chatterjee, K.; Lin-Gibson, S.; Wallace, W.E.; Parekh, S.H.; Lee, Y.J.; Cicerone, M.T.; Young, M.F.; Simon, C.G. The Effect of 3D Hydrogel Scaffold Modulus on Osteoblast Differentiation and Mineralization Revealed by Combinatorial Screening. Biomaterials 2010, 31, 5051–5062.
- Guda, T.; Oh, S.; Appleford, M.R.; Ong, J.L. Bilayer Hydroxyapatite Scaffolds for Maxillofacial Bone Tissue Engineering. Int. J. Oral Maxillofac. Implants 2012, 27, 288–294.
- Gothard, D.; Smith, E.L.; Kanczler, J.M.; Black, C.R.; Wells, J.A.; Roberts, C.A.; White, L.J.; Qutachi, O.; Peto, H.; Rashidi, H.; et al. In Vivo Assessment of Bone Regeneration in Alginate/Bone ECM Hydrogels with Incorporated Skeletal Stem Cells and Single Growth Factors. PLoS ONE 2015, 10, e0145080.
- Celikkin, N.; Mastrogiacomo, S.; Walboomers, X.; Swieszkowski, W. Enhancing X-Ray Attenuation of 3D Printed Gelatin Methacrylate (GelMA) Hydrogels Utilizing Gold Nanoparticles for Bone Tissue Engineering Applications. Polymers 2019, 11, 367.
- Pan, T.; Song, W.; Cao, X.; Wang, Y. 3D Bioplotting of Gelatin/Alginate Scaffolds for Tissue Engineering: Influence of Crosslinking Degree and Pore Architecture on Physicochemical Properties. J. Mater. Sci. Technol. 2016, 32, 889–900.
- Spiller, K.L.; Holloway, J.L.; Gribb, M.E.; Lowman, A.M. Design of Semi-Degradable Hydrogels Based on Poly(Vinyl Alcohol) and Poly(Lactic-Co-Glycolic Acid) for Cartilage Tissue Engineering. J. Tissue Eng. Regen. Med. 2011, 5, 636–647.
- Behravesh, E.E.; Timmer, M.D.; Lemoine, J.J.; Liebschner, M.A.K.; Mikos, A.G. Evaluation of the in Vitro Degradation of Macroporous Hydrogels Using Gravimetry, Confined Compression Testing, and Microcomputed Tomography. Biomacromolecules 2002, 3, 1263–1270.
- Hedberg, E.L.; Shih, C.K.; Lemoine, J.J.; Timmer, M.D.; Liebschner, M.A.; Jansen, J.A.; Mikos, A.G. In Vitro Degradation of Porous Poly(Propylene Fumarate)/Poly(Dl-Lactic-Co-Glycolic Acid) Composite Scaffolds. Biomaterials 2005, 26, 3215–3225.
- Shi, M.; Kretlow, J.D.; Nguyen, A.; Young, S.; Scott Baggett, L.; Wong, M.E.; Kurtis Kasper, F.; Mikos, A.G. Antibiotic-Releasing Porous Polymethylmethacrylate Constructs for Osseous Space Maintenance and Infection Control. Biomaterials 2010, 31, 4146–4156.
- Vásárhelyi, L.; Kónya, Z.; Kukovecz, Á.; Vajtai, R. Microcomputed Tomography–Based Characterization of Advanced Materials: A Review. Mater. Today Adv. 2020, 8, 100084.
- Dubruel, P.; Unger, R.; van Vlierberghe, S.; Cnudde, V.; Jacobs, P.J.S.; Schacht, E.; Kirkpatrick, C.J. Porous Gelatin Hydrogels: 2. In Vitro Cell Interaction Study. Biomacromolecules 2007, 8, 338–344.
- Offeddu, G.S.; Ashworth, J.C.; Cameron, R.E.; Oyen, M.L. Structural Determinants of Hydration, Mechanics and Fluid Flow in Freeze-Dried Collagen Scaffolds. Acta Biomater. 2016, 41, 193–203.
- Wu, X.; Wang, X.; Chen, X.; Yang, X.; Ma, Q.; Xu, G.; Yu, L.; Ding, J. Injectable and Thermosensitive Hydrogels Mediating a Universal Macromolecular Contrast Agent with Radiopacity for Noninvasive Imaging of Deep Tissues. Bioact. Mater. 2021, 6, 4717–4728.
- Patrick, P.S.; Bear, J.C.; Fitzke, H.E.; Zaw-Thin, M.; Parkin, I.P.; Lythgoe, M.F.; Kalber, T.L.; Stuckey, D.J. Radio-Metal Cross-Linking of Alginate Hydrogels for Non-Invasive in Vivo Imaging. Biomaterials 2020, 243, 119930.
- Faraj, K.A.; Cuijpers, V.M.J.I.; Wismans, R.G.; Walboomers, X.F.; Jansen, J.A.; van Kuppevelt, T.H.; Daamen, W.F. Micro-Computed Tomographical Imaging of Soft Biological Materials Using Contrast Techniques. Tissue Eng. Part C Methods 2009, 15, 493–499.
- Lichtman, J.W.; Conchello, J.-A. Fluorescence Microscopy. Nat. Methods 2005, 2, 910–919.
- Hickey, S.M.; Ung, B.; Bader, C.; Brooks, R.; Lazniewska, J.; Johnson, I.R.D.; Sorvina, A.; Logan, J.; Martini, C.; Moore, C.R.; et al. Fluorescence Microscopy—An Outline of Hardware, Biological Handling, and Fluorophore Considerations. Cells 2021, 11, 35.
- Rai, V.; Dey, N. The Basics of Confocal Microscopy. In Laser Scanning, Theory and Applications; Wang, C.-C., Ed.; InTech: London, UK, 2011; ISBN 978-953-307-205-0.
- Moshkov, A. Confocal Laser Scanning Microscopy of Living Cells. In Fluorescence Methods for Investigation of Living Cells and Microorganisms; Grigoryeva, N., Ed.; IntechOpen: London, UK, 2020; ISBN 978-1-83968-039-7.
- Sapudom, J.; Rubner, S.; Martin, S.; Kurth, T.; Riedel, S.; Mierke, C.T.; Pompe, T. The Phenotype of Cancer Cell Invasion Controlled by Fibril Diameter and Pore Size of 3D Collagen Networks. Biomaterials 2015, 52, 367–375.
- Bagnaninchi, P.O.; Yang, Y.; Zghoul, N.; Maffulli, N.; Wang, R.K.; El Haj, A.J. Chitosan Microchannel Scaffolds for Tendon Tissue Engineering Characterized Using Optical Coherence Tomography. Tissue Eng. 2007, 13, 323–331.
- Hayn, A.; Fischer, T.; Mierke, C.T. Inhomogeneities in 3D Collagen Matrices Impact Matrix Mechanics and Cancer Cell Migration. Front. Cell Dev. Biol. 2020, 8, 593879.
- Fischer, T.; Wilharm, N.; Hayn, A.; Mierke, C.T. Matrix and Cellular Mechanical Properties Are the Driving Factors for Facilitating Human Cancer Cell Motility into 3D Engineered Matrices. Converg. Sci. Phys. Oncol. 2017, 3, 044003.
- Fischer, T.; Hayn, A.; Mierke, C.T. Fast and Reliable Advanced Two-Step Pore-Size Analysis of Biomimetic 3D Extracellular Matrix Scaffolds. Sci. Rep. 2019, 9, 8352.
- Fischer, T.; Hayn, A.; Mierke, C.T. Effect of Nuclear Stiffness on Cell Mechanics and Migration of Human Breast Cancer Cells. Front. Cell Dev. Biol. 2020, 8, 393.
- Bancelin, S.; Aimé, C.; Gusachenko, I.; Kowalczuk, L.; Latour, G.; Coradin, T.; Schanne-Klein, M.-C. Determination of Collagen Fibril Size via Absolute Measurements of Second-Harmonic Generation Signals. Nat. Commun. 2014, 5, 4920.
- Doyle, A.D.; Carvajal, N.; Jin, A.; Matsumoto, K.; Yamada, K.M. Local 3D Matrix Microenvironment Regulates Cell Migration through Spatiotemporal Dynamics of Contractility-Dependent Adhesions. Nat. Commun. 2015, 6, 8720.
More
Information
Subjects:
Cell & Tissue Engineering
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.9K
Revisions:
3 times
(View History)
Update Date:
31 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No