 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonios Lazaridis | -- | 2785 | 2022-08-26 22:21:47 | | | |
| 2 | Dean Liu | Meta information modification | 2785 | 2022-08-29 02:58:11 | | |
Video Upload Options
Vascular aging, characterized by structural and functional alterations of the vascular wall, is a hallmark of aging and is tightly related to the development of cardiovascular mortality and age-associated vascular pathologies.
1. Introduction
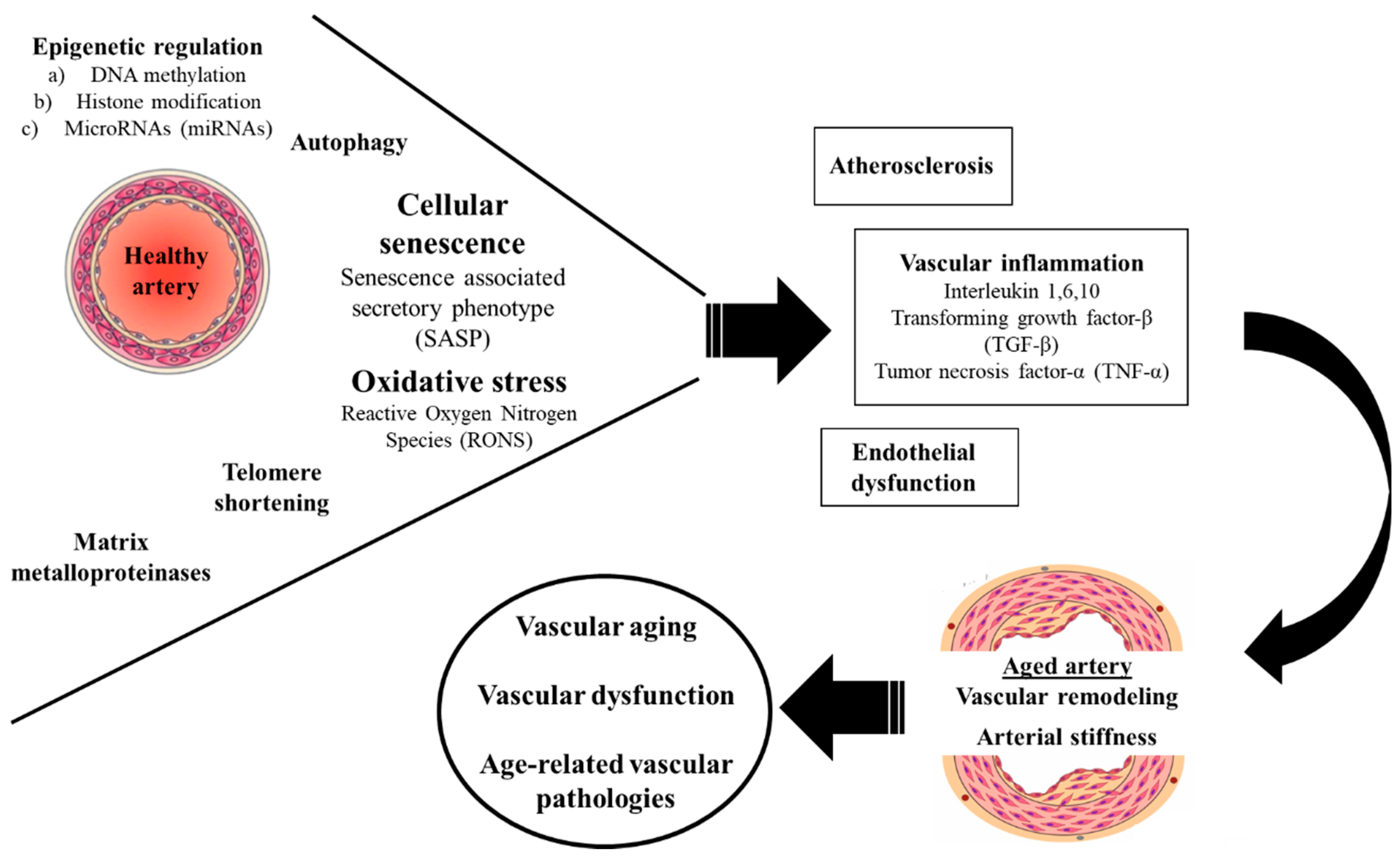
Figure 1. Molecular mechanisms of vascular aging.
2. Oxidative Stress
3. Inflammation
4. Extracellular Matrix Metallorproteinases
The healthy vasculature comprises of the ECs, VSMCs and the ECM, all of which are susceptible to damage or disruption during aging [27]. The ECM is composed of structural proteins such as collagens and elastin that tether VSMCs together, provide structural support, and regulate the mechanical function of the vessel [28]. Disruption of ECM integrity by MMPs greatly changes its composition and substantially impacts vascular homeostasis during aging through structural and functional changes of the vessel wall.
MMPs belong to a family of zinc dependent endopeptidases and are mainly extracellular proteins, even though some members are also found intracellularly and may act on intracellular proteins. Several MMPs have been implicated in age related pathologies including MMP-2,3,7,9 [29][30][31][32]. The contribution of MMPs in vascular aging has been further corroborated by the observations of vascular impact upon MMP inhibition. It has been shown that tissue inhibitors of MMPs (TIMPs) including four molecules (TIMP-1,-2,-3,-4), reversibly inhibit the proteolytic activity of activated MMPs and an imbalance of MMPs and TIMPs has been implicated in hypertension, atherosclerotic plaque formation and aortic aneurysm formation in several experimental models [33]. More specifically, it has been demonstrated that overexpression of TIMP-1 by gene transfer can reduce balloon injury-induced intimal formation while TIMP-3 deficiency enhances inflammation and aggravates atherosclerosis in ApoE-knockout mice [34]. In addition to this, TIMP-3 has been demonstrated to mediate the inhibitory effect of interleukin-32α on endothelial inflammation, smooth muscle cell activation, and development of atherosclerosis [35]. Similar effects have been demonstrated for TIMP-2, and TIMP-4, mainly through mechanisms of VSMC migration and apoptosis [36][37]. Furthermore, TIMP-1 appears to protect against aortic aneurysm formation and rupture in rat models since its overexpression prevents elastin degradation. Similarly, in response to AngII, TIMP-3 gene deletion in non-atherosclerotic mice has been shown to trigger adverse remodeling of the abdominal aorta [38].
5. Epigenetic Regulation
DNA methylation
DNA methylation is a dynamically reversible process that modifies the genome function through the addition of methyl groups to cytosine in order to form 5-methyl-cytosine (5mC) and it is regulated by DNA methyltransferases (DNMT1, DNMT3A and DNMT3B) and demethyltransferases. In general, DNA methylation and hypermethylation inhibit gene expression either by recruiting proteins which are implicated in gene repression or by impeding the binding of transcription factors to DNA [39]. On the other hand, DNA demethylation or hypomethylation preserves gene expression, although at a cost, since it can initiate transcription at an incorrect gene region or even exhibit high transcriptional activity in normally silent sites. Therefore, hypomethylation may cause structural changes, chromosome instability and expression of potentially harmful genes [40]. Accumulating evidence has identified several genes which are regulated through different levels of DNA methylation and are involved in the development of vascular aging by modulating the function of several vascular cells such as ECs, VSMCs and macrophages [41].
Histone modification
Histone modification is a process during which chromatin structure and function as well as gene expression, transcription and repair are regulated. Similarly, post translational modifications are also determined by this mechanism [42]. This regulation is enabled by the interaction between histone proteins and DNA. The mechanisms in charge of histone modification include acetylation, methylation, phosphorylation and ubiquitation. Accordingly, the main enzymes involved are histone acetyl transferases (HATs), deacetylases (HDACs), methyltransferases (HMTs) and demethylases (HDMs) [43]. Among all HDACs, sirtuins are the most widely studied and Sirt1 is the best characterized member in relation to vascular aging.
Sirt1 is systematically expressed at vascular level by several cells including ECs, monocytes/macrophages and VSMCs and is implicated in deacetylation of several transcriptional factors, co-regulatory proteins and enzymes like peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1a), NF-κB, eNOS, FOXO, p53, p300/CBP, H3H9 and H3K56 [44][130]. Overall, previous studies have revealed in detail the protective role of Sirt1 against vascular aging through abundant beneficial effects in the structural and functional homeostasis of the vasculature [45]. A detailed summary of the mechanisms through which Sirt1 protects against vascular aging is depicted in Table 1.
|
Table 1. Beneficial mechanisms through which SIRT1 upregulation protects against vascular aging |
|
Recruitment of EC migration |
|
Delay of the aging and dysfunction of EPCs |
|
Inhibition of aging of ECs by binding the PAI-1 promoter and by deacetylation of histone H4K16 |
|
Promotion of endothelial KLF2 expression which enables transition of ECs to a „vaso-protective“ state |
|
Mitigation of hyperglycaemia-induced endothelial dysfunction due to ROS production by inhibiting vascular p66Shc gene transcription |
|
Alleviation of oxidative stress and inflammation by the inhibition of NF-κB signalling pathway |
|
Activation of eNOS and promotion of NO production by the deacetylation of eNOS on Lys496 and Lys506 |
|
Reduction of COX-2 expression through downregulation of transcription factor AP-1 in macrophages |
|
Reduction of arterial remodelling and stiffness by the alleviation of oxidative stress in VSMCs |
|
Deacetylation and activation of the FOXO 1, 3, and 4 transcription factors leading to the expression of several antioxidant genes |
AP-1: activator protein-1; COX-2: cyclooxygenase-2; ECs: endothelial cells; eNOS: endothelial nitric oxide synthase; EPCs: endothelial progenitor cells; FOXO: forkhead fox; HUVECs: human umbilical vein endothelial cells; KLF2: Kruppel-like factor 2; NF-κB: nuclear factor kappa B; NO: nitric oxide; PAI-1: plasminogen activator inhibitor-1; PARP: Poly (ADP-ribose) polymerase; ROS: reactive oxygen species; VSMCs: vascular smooth muscle cells |
Non-coding RNAs
The non-coding RNAs (ncRNAs) represent RNA molecules that lack protein coding potential and are divided, according to their nucleotide content, into short or small ncRNAs (< 200 nucleotides) and long ncRNAs (> 200 nucleotides). Furthermore, microRNAs (miRNAs) (21–25 nucleotides) belong to the short ncRNAs and are the most extensively studied member while recently discovered circular RNAs (300–500 nucleotides) pertain to the long ncRNAs. The ncRNAs play a significant role in the post-transcriptional genetic regulation. In particular, miRNAs negatively regulate gene expression by binding a target mRNA and inducing its degradation or by inhibiting its translation [46]. Increasing evidence has shown that miRNAs have a considerable impact on various molecular mechanisms related to vascular function and aging (Table 2).
|
Table 2. Major miRNAs and their involvement in vascular aging |
|
|
miR-10A |
Propagation of senescence of endothelial progenitor cells through suppression of the high-mobility group A2 molecule |
|
miR-21 |
Propagation of senescence of endothelial progenitor cells through suppression of the high-mobility group A2 molecule |
|
miR-22 |
Inhibition of VSMC proliferation and migration and neointima formation |
|
miR-34a |
Suppression of EC proliferation and promotion of EC senescence in part through Sirt1 inhibition Impairment of EPC-mediated angiogenesis through suppression of silent information regulator 1 |
|
miR-126 |
Reduction of endothelial inflammation through inhibition of VCAM-1 expression |
|
miR-128 |
Reduction of VSMC proliferation, migration, and contractility |
|
miR-143 |
Inhibition of VSMC proliferation through targeting the transcription factor Elk-1 |
|
miR-145 |
Inhibition of VSMC proliferation through targeting the transcription factor myocardin |
|
miR-146a |
Promotion of VSMC proliferation and vascular neointimal hyperplasia through targeting KLF4 |
|
miR-155 |
Promotion of atherosclerosis through repression of macrophage BCL6 expression Endothelial dysfunction and vasoconstriction through downregulation of eNOS and sGCβ1 expression |
|
miR-217 |
Acceleration of EC senescence, endothelial dysfunction and development of atherosclerosis through Sirt1 downregulation |
BCL6: B-cell lymphoma 6 protein; BP: blood pressure; EC: endothelial cell; eNOS: endothelial nitric oxide synthase; KLF4: Krüppel-like factor 4; sGCβ1: soluble guanylyl cyclase β1; VCAM-1: vascular cell adhesion molecule-1; VEGF: vascular endothelial growth factor; VSMC: vascular smooth muscle cells |
|
6. Telomere Shortening
Telomeres are non-coding DNA structures consisting of a repetitive hexanucleotide DNA sequence (TTAGGG) found in the terminal loops, where they cap and stabilize the physical ends of eukaryotic chromosomes [47]. While aging telomeres shorten with each successive cell division, however, below a critical length they induce the DNA damage response (DDR), eventually leading to replicative senescence and the end of cellular proliferation. Actually, telomere shortening or attrition constitutes a major triggering factor of senescence leading to vascular aging and cardiovascular disease [48].
Abundant experimental data have linked telomere shortening with the development of endothelial dysfunction and atherogenesis [49]. Furthermore, clinical data have highlighted the association of telomere length with arterial stiffness and atherosclerotic burden across different age and cardiovascular risk populations but also healthy individuals; hence, shorter telomeres have been associated with increased aortic pulse wave velocity [50], pulse pressure [51] and carotid IMT [52]. In addition, leukocyte telomere length is decreased in patients with various cardiovascular disease phenotypes including heart failure, myocardial infarction [53] and atherosclerotic hypertensive disease [54]. More importantly, telomere length has been closely correlated with cardiovascular risk in several large cohorts and meta-analyses
7. Cellular Senescence
Cellular senescence is a state of a durable, irreversible cell-cycle arrest of previously replication-competent cells [55] which plays a dual role in physiology and disease [56]. Senescence has been recognized as a central hallmark of aging since most of its stimuli including telomere attrition, mitochondrial dysfunction, oncogene activation, and DNA damage, are primary drivers of the process. Importantly, senescence is also by itself a key driver of vascular dysfunction and aging by mediating endothelial dysfunction, inflammation, and atherosclerosis [57]. More specifically, early in vitro observations have shown that induction of senescence in human aortic ECs reduces levels of NO and increases expression ICAM-1 [58].
Τhe senescence-associated secretory phenotype (SASP) consists of a plethora of factors produced by the senescent cells including pro-inflammatory cytokines and chemokines, growth modulators, angiogenic factors, and MMPs that can induce inflammation, stem cell dysfunction, immunity activation, apoptosis and further trigger senescence in neighboring cells [57][59]. The net result is a state of persistent chronic inflammation, known as inflammaging which is tightly associated with multiple age-related phenotypes [60]. In close association with this, experimental data have documented considerable accumulation of senescent VSMCs and ECs in human atherosclerotic lesions that persistently express key SASP factors [61]; hence installing a highly inflammatory and pro-atherogenic environment which contributes to the progression of atherosclerosis [62].
Additionally, strong evidence advocating the contribution of senescence to vascular aging comes from preclinical studies investigating pharmacologic agents which lead to the ‘‘senolytic’’ clearance of senescent cells and attenuation of inflammation[63]. Finally, alternative pharmacologic approaches have emerged including drugs that prevent the progression of cell senescence without inducing the death of senescent cells (senomorphic drugs) such as SASP inhibitors [64].
8. Autophagy
Autophagy is a highly selective physiological process by which cells encapsulate and deliver their macromolecular components such as proteins and organelles to lysosomes for subsequent degradation [65]. Importantly, with aging, there is a progressive reduction in the autophagic activity across several species and model systems [66], which has been further associated vascular dysfunction, accelerated aging, and several age-related vascular diseases [67].
Data from aged mice and human subjects have shown that compromised autophagy of ECs is associated with a markedly blunted endothelial-dependent vasodilative response [68]. Coincident with this effect, it has been demonstrated that loss of autophagy promotes an increase in endothelial ROS and inflammatory cytokines, hence suggesting that autophagy may regulate vascular homeostasis, in part, through a NO-dependent pathway [69].
Contrary to the harmful effects of impaired autophagy, several lines of evidence have corroborated that induction of autophagy has a protective effect on vascular homeostasis. The lifestyle modification of caloric restriction, which is the most effective strategy to induce autophagy so far, has been shown to improve vascular function in both rodent models and human subjects by intervening in crucial regulatory pathways including the deacetylase Sirt1, the AMP-activated protein kinase (AMPK), and the mammalian target of rapamycin (mTOR) [66].Regarding vascular aging, it has been shown that long-term caloric restriction in mice prevents the age-related declines in endothelial function and increases in large elastic artery stiffness and these effects are related to reduced oxidative stress [70]. Likewise, short-term (i.e., 3–8 weeks) caloric restriction also reverses the age-related vascular dysfunction in old mice. Additionally, in humans, caloric restriction-based weight loss in overweight and obese middle-aged and older adults has been shown to improve macrovascular and microvascular endothelial function and large elastic artery stiffness [71].
9. Conclusion
Vascular aging and the associated changes in the vascular wall represent a certain hallmark of the aging process that are irrefragably related to increased cardiovascular mortality and the development of several age-related pathologies. Accumulating evidence over the last years has called attention on several complex molecular pathways implicated in the pathophysiology of vascular aging which are a matter of intense investigation. Among them, oxidative stress, arteriosclerosis, vascular inflammation and the related endothelial dysfunction, seem to represent the common denominator that accelerates vascular ageing and stiffening of the arteries. Within this conceptual framework, a deeper understanding of these highly sophisticated biological processes is warranted in order to develop certain therapeutic targets and facilitate future interventions aiming to improve human health span and longevity.
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217.
- Freedman, V.A.; Martin, L.G.; Schoeni, R.F. Recent Trends in Disability and Functioning among Older Adults in the United States: A Systematic Review. JAMA 2002, 288, 3137–3146.
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part I: Aging Arteries: A “Set up” for Vascular Disease. Circulation 2003, 107, 139–146.
- Paneni, F.; Diaz Cañestro, C.; Libby, P.; Lüscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding It at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967.
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61.
- Tauzin, L. Alterations in Viscoelastic Properties Following Premature Birth May Lead to Hypertension and Cardiovascular Disease Development in Later Life. Acta Paediatr. Int. J. Paediatr. 2015, 104, 19–26.
- Martyn, C.N.; Greenwald, S.E. Impaired Synthesis of Elastin in Walls of Aorta and Large Conduit Arteries during Early Development as an Initiating Event in Pathogenesis of Systemic Hypertension. Lancet 1997, 350, 953–955.
- Martin, H.; Hu, J.; Gennser, G.; Norman, M. Impaired Endothelial Function and Increased Carotid Stiffness in 9-Year-Old Children with Low Birthweight. Circulation 2000, 102, 2739–2744.
- Brodszki, J.; Länne, T.; Maršál, K.; Ley, D. Impaired Vascular Growth in Late Adolescence after Intrauterine Growth Restriction. Circulation 2005, 111, 2623–2628.
- Xu, X.; Wang, B.; Ren, C.; Hu, J.; Greenberg, D.A.; Chen, T.; Xie, L.; Jin, K. Age-Related Impairment of Vascular Structure and Functions. Aging Dis. 2017, 8, 590–610.
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848.
- Laina, A.; Stellos, K.; Stamatelopoulos, K. Vascular Ageing: Underlying Mechanisms and Clinical Implications. Exp. Gerontol. 2018, 109, 16–30.
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867.
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748.
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-morte, D.; Testa, G.; Cacciatore, F.; Bonaduce, D.; Abete, P. Oxidative Stress and Diseases. Oxidative Stress Dis. 2018, 47, 770–773.
- Martínez-Revelles, S.; García-Redondo, A.B.; Avendaño, M.S.; Varona, S.; Palao, T.; Orriols, M.; Roque, F.R.; Fortuño, A.; Touyz, R.M.; Martínez-González, J.; et al. Lysyl Oxidase Induces Vascular Oxidative Stress and Contributes to Arterial Stiffness and Abnormal Elastin Structure in Hypertension: Role of P38MAPK. Antioxid. Redox Signal. 2017, 27, 379–397.
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1–11.
- Durackova, Z. Systems Biology of Free Radicals and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9783642300.
- Alberts-Grill, N.; Denning, T.L.; Rezvan, A.; Jo, H. The Role of the Vascular Dendritic Cell Network in Atherosclerosis. Am. J. Physiol. Cell Physiol. 2013, 305, C1–C21.
- Zuliani, G.; Morieri, M.L.; Volpato, S.; Maggio, M.; Cherubini, A.; Francesconi, D.; Bandinelli, S.; Paolisso, G.; Guralnik, J.M.; Ferrucci, L. Insulin Resistance and Systemic Inflammation, but Not Metabolic Syndrome Phenotype, Predict 9 Years Mortality in Older Adults. Atherosclerosis 2014, 235, 538–545.
- Ferrucci, L.; Harris, T.B.; Guralnik, J.M.; Tracy, R.P.; Corti, M.-C.; Cohen, H.J.; Penninx, B.; Pahor, M.; Wallace, R.; Havlik, R.J. Serum IL-6 Level and the Development of Disability in Older Persons. Am. Geriatr. Soc. 1999, 47, 639–646.
- Fabbri, E.; An, Y.; Zoli, M.; Simonsick, E.M.; Guralnik, J.M.; Bandinelli, S.; Boyd, C.M.; Ferrucci, L. Aging and the Burden of Multimorbidity: Associations with Inflammatory and Anabolic Hormonal Biomarkers. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 63–70.
- Soysal, P.; Stubbs, B.; Lucato, P.; Luchini, C.; Solmi, M.; Peluso, R.; Sergi, G.; Isik, A.T.; Manzato, E.; Maggi, S.; et al. Inflammation and Frailty in the Elderly: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2016, 31, 1–8.
- Montero, I.; Orbe, J.; Varo, N.; Beloqui, O.; Monreal, J.I.; Rodréguez, J.A.; Díez, J.; Libby, P.; Páramo, J.A. C-Reactive Protein Induces Matrix Metalloproteinase-1 and -10 in Human Endothelial Cells: Implications for Clinical and Subclinical Atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, 1369–1378.
- Scicali, R.; Di Pino, A.; Urbano, F.; Ferrara, V.; Marchisello, S.; Di Mauro, S.; Scamporrino, A.; Filippello, A.; Piro, S.; Rabuazzo, A.M.; et al. Analysis of S100A12 Plasma Levels in Hyperlipidemic Subjects with or without Familial Hypercholesterolemia. Acta Diabetol. 2019, 56, 899–906.
- Wang, W.; Deng, Z.; Li, L.; Li, J.; Jin, X. Association of Hyper-Sensitive C-Reactive Protein with Arterial Stiffness and Endothelial Function in Patients with Hyperlipidemia. Int. J. Clin. Exp. Med. 2016, 9, 23416–23424.
- Patrick Lacolley; Veronique Regnault; Alberto P Avolio; Smooth muscle cell and arterial aging: basic and clinical aspects. Cardiovascular Research 2018, 114, 513-528, 10.1093/cvr/cvy009.
- Kirsty Foote; Martin R. Bennett; Molecular insights into vascular aging. Aging 2018, 10, 3647-3649, 10.18632/aging.101697.
- Adam Harvey; Augusto C. Montezano; Rheure Alves Lopes; Francisco Rios; Rhian M. Touyz; Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Canadian Journal of Cardiology 2016, 32, 659-668, 10.1016/j.cjca.2016.02.070.
- Jason L. Johnson; Sarah J. George; Andrew C. Newby; Christopher L. Jackson; Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proceedings of the National Academy of Sciences 2005, 102, 15575-15580, 10.1073/pnas.0506201102.
- Ji Yoon Park; Jun Hyoung Park; Wookju Jang; In-Kwan Hwang; In Ja Kim; Hwa-Jung Kim; Kyung-Hyun Cho; Seung-Taek Lee; Apolipoprotein A-IV is a novel substrate for matrix metalloproteinases. The Journal of Biochemistry 2011, 151, 291-298, 10.1093/jb/mvr137.
- Jon M. Florence; Agnieszka Krupa; Laela M. Booshehri; Timothy C. Allen; Anna K. Kurdowska; Metalloproteinase-9 contributes to endothelial dysfunction in atherosclerosis via protease activated receptor-1. PLOS ONE 2017, 12, e0171427, 10.1371/journal.pone.0171427.
- Utpal Sen; Regulation and involvement of matrix metalloproteinases in vascular diseases. Frontiers in Bioscience 2016, 21, 89-118, 10.2741/4378.
- Robert Stöhr; Michele Cavalera; Stefano Menini; Maria Mavilio; Viviana Casagrande; Claudia Rossi; Andrea Urbani; Marina Cardellini; Giuseppe Pugliese; Rossella Menghini; et al.Massimo Federici Loss of TIMP3 exacerbates atherosclerosis in ApoE null mice. Atherosclerosis 2014, 235, 438-443, 10.1016/j.atherosclerosis.2014.05.946.
- Dong Ju Son; Yu Yeon Jung; Young Sik Seo; Heonyong Park; Dong Hun Lee; Sanghyeon Kim; Yoon-Seok Roh; Sang Bae Han; Do Young Yoon; Jin Tae Hong; et al. Interleukin-32α Inhibits Endothelial Inflammation, Vascular Smooth Muscle Cell Activation, and Atherosclerosis by Upregulating Timp3 and Reck through suppressing microRNA-205 Biogenesis. Theranostics 2017, 7, 2186-2203, 10.7150/thno.18407.
- Jason L. Johnson; Andrew H. Baker; Kazuhiro Oka; Lawrence Chan; Andrew C. Newby; Christopher L. Jackson; Sarah J. George; Suppression of Atherosclerotic Plaque Progression and Instability by Tissue Inhibitor of Metalloproteinase-2. Circulation 2006, 113, 2435-2444, 10.1161/circulationaha.106.613281.
- Joseph D. Raffetto; Raouf A. Khalil; Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochemical Pharmacology 2008, 75, 346-359, 10.1016/j.bcp.2007.07.004.
- Dong Fan; Zamaneh Kassiri; Biology of Tissue Inhibitor of Metalloproteinase 3 (TIMP3), and Its Therapeutic Implications in Cardiovascular Pathology. Frontiers in Physiology 2020, 11, 1-16, 10.3389/fphys.2020.00661.
- Lisa D Moore; Thuc Le; Guoping Fan; DNA Methylation and Its Basic Function. Neuropsychopharmacology 2012, 38, 23-38, 10.1038/npp.2012.112.
- Samira Tabaei; Seyyedeh Samaneh Tabaee; DNA methylation abnormalities in atherosclerosis. Artificial Cells, Nanomedicine, and Biotechnology 2019, 47, 2031-2041, 10.1080/21691401.2019.1617724.
- Hui Xu; Shuang Li; You-Shuo Liu; Roles and Mechanisms of DNA Methylation in Vascular Aging and Related Diseases. Frontiers in Cell and Developmental Biology 2021, 9, 699374, 10.3389/fcell.2021.699374.
- Brian D Strahl; C. David Allis; The language of covalent histone modifications. Nature Cell Biology 2000, 403, 41-45, 10.1038/47412.
- Guo-Hua Ding; Dan-Di Guo; Yang Guan; Chun-Yu Chi; Bao-Dong Liu; Changes of DNA methylation of Isoetes sinensis under Pb and Cd stress. Environmental Science and Pollution Research 2018, 26, 3428-3435, 10.1007/s11356-018-3864-3.
- Marta Halasa; Kamila Adamczuk; Grzegorz Adamczuk; Syeda Afshan; Andrzej Stepulak; Marek Cybulski; Anna Wawruszak; Deacetylation of Transcription Factors in Carcinogenesis. International Journal of Molecular Sciences 2021, 22, 11810, 10.3390/ijms222111810.
- Munehiro Kitada; Yoshio Ogura; Daisuke Koya; The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290-2307, 10.18632/aging.101068.
- Julia Beermann; Maria-Teresa Piccoli; Janika Viereck; Thomas Thum; Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiological Reviews 2016, 96, 1297-1325, 10.1152/physrev.00041.2015.
- Anna K. Uryga; Martin Bennett; Ageing induced vascular smooth muscle cell senescence in atherosclerosis. The Journal of Physiology 2015, 594, 2115-2124, 10.1113/jp270923.
- Yu Liu; Samuel I. Bloom; Anthony J. Donato; The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2018, 26, e12487-e12487, 10.1111/micc.12487.
- Charles Matthews; Isabelle Gorenne; Stephen Scott; Nicola Figg; Peter Kirkpatrick; Andrew Ritchie; Martin Goddard; Martin Bennett; Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis. Circulation Research 2006, 99, 156-164, 10.1161/01.res.0000233315.38086.bc.
- Barry J. McDonnell; Yasmin; Lee Butcher; John R. Cockcroft; Ian B. Wilkinson; Jorge Erusalimsky; Carmel M. McEniery; The age-dependent association between aortic pulse wave velocity and telomere length. The Journal of Physiology 2017, 595, 1627-1635, 10.1113/jp273689.
- Elisabeth Jeanclos; Nicholas J. Schork; Kirsten Ohm Kyvik; Masayuki Kimura; Joan H. Skurnick; Abraham Aviv; Telomere Length Inversely Correlates With Pulse Pressure and Is Highly Familial. Hypertension 2000, 36, 195-200, 10.1161/01.hyp.36.2.195.
- Yun-Ying Wang; Ai-Fang Chen; Hua-Zhang Wang; Li-Yan Xie; Ke-Xu Sui; Qi-Yi Zhang; Association of shorter mean telomere length with large artery stiffness in patients with coronary heart disease. The Aging Male 2010, 14, 27-32, 10.3109/13685538.2010.529196.
- Pim van der Harst; Gerrit van der Steege; Rudolf A. de Boer; Adriaan A. Voors; Alistair Hall; Marcel J. Mulder; Wiek van Gilst; Dirk J. van Veldhuisen; Telomere Length of Circulating Leukocytes Is Decreased in Patients With Chronic Heart Failure. Journal of the American College of Cardiology 2007, 49, 1459-1464, 10.1016/j.jacc.2007.01.027.
- Athanase Benetos; Jeffrey P. Gardner; Mahmoud Zureik; Carlos Labat; Lu Xiaobin; Chris Adamopoulos; Mohamed Temmar; Kathryn E. Bean; Frédérique Thomas; Abraham Aviv; et al. Short Telomeres Are Associated With Increased Carotid Atherosclerosis in Hypertensive Subjects. Hypertension 2004, 43, 182-185, 10.1161/01.hyp.0000113081.42868.f4.
- Vassilis Gorgoulis; Peter D. Adams; Andrea Alimonti; Dorothy Bennett; Oliver Bischof; Cleo Bishop; Judith Campisi; Manuel Collado; Konstantinos Evangelou; Gerardo Ferbeyre; et al.Jesus GilEiji HaraValery KrizhanovskyDiana JurkAndrea B. MaierMasashi NaritaLaura NiedernhoferJoão F. PassosPaul D. RobbinsClemens A. SchmittJohn SedivyKonstantinos VougasThomas von ZglinickiDaohong ZhouManuel SerranoMarco DeMaria Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813-827, 10.1016/j.cell.2019.10.005.
- Shenghui He; Norman E. Sharpless; Senescence in Health and Disease. Cell 2017, 169, 1000-1011, 10.1016/j.cell.2017.05.015.
- Raffaella Di Micco; Valery Krizhanovsky; Darren Baker; Fabrizio D’Adda di Fagagna; Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nature Reviews Molecular Cell Biology 2020, 22, 75-95, 10.1038/s41580-020-00314-w.
- Tohru Minamino; Hideaki Miyauchi; Toshihiko Yoshida; Yasuo Ishida; Hideo Yoshida; Issei Komuro; Endothelial Cell Senescence in Human Atherosclerosis. Circulation 2002, 105, 1541-1544, 10.1161/01.cir.0000013836.85741.17.
- Juan Carlos Acosta; Ana Banito; Torsten Wuestefeld; Athena Georgilis; Peggy Janich; Jennifer P. Morton; Dimitris Athineos; Tae-Won Kang; Felix Lasitschka; Mindaugas Andrulis; et al.Gloria PascualKelly J. MorrisSadaf KhanHong JinGopuraja DharmalingamBram SnijdersThomas CarrollDavid CapperCatrin PritchardGareth J. InmanThomas LongerichOwen J. SansomSalvador Aznar BenitahLars ZenderJesús Gil A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nature Cell Biology 2013, 15, 978-990, 10.1038/ncb2784.
- Claudio Franceschi; Judith Campisi; Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. The Journals of Gerontology: Series A 2014, 69, S4-S9, 10.1093/gerona/glu057.
- Lee Ann Campbell; Cho-Chou Kuo; Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Seminars in Respiratory Infections 2003, 18, 48-54, 10.1053/srin.2003.50006.
- Bennett G. Childs; Darren J. Baker; Tobias Wijshake; Cheryl A. Conover; Judith Campisi; Jan M. Van Deursen; Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472-477, 10.1126/science.aaf6659.
- James L. Kirkland; Tamara Tchkonia; Cellular Senescence: A Translational Perspective. eBioMedicine 2017, 21, 21-28, 10.1016/j.ebiom.2017.04.013.
- Lisa A. Lesniewski; Douglas R. Seals; Ashley Walker; Grant D. Henson; Mark W. Blimline; Daniel W. Trott; Gary C. Bosshardt; Thomas J. LaRocca; Brooke R. Lawson; Melanie C. Zigler; et al.Anthony J. Donato Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 2016, 16, 17-26, 10.1111/acel.12524.
- Zhangyuan Yin; Clarence Pascual; Daniel J. Klionsky; Autophagy: machinery and regulation. Microbial Cell 2016, 3, 588-596, 10.15698/mic2016.12.546.
- Mahmoud Abdellatif; Simon Sedej; Didac Carmona-Gutierrez; Frank Madeo; Guido Kroemer; Autophagy in Cardiovascular Aging. Circulation Research 2018, 123, 803-824, 10.1161/circresaha.118.312208.
- Samuel C. Nussenzweig; Subodh Verma; Toren Finkel; The Role of Autophagy in Vascular Biology. Circulation Research 2015, 116, 480-488, 10.1161/circresaha.116.303805.
- Thomas J. LaRocca; Grant D. Henson; Andrew Thorburn; Amy L. Sindler; Gary L. Pierce; Douglas R. Seals; Translational evidence that impaired autophagy contributes to arterial ageing. The Journal of Physiology 2012, 590, 3305-3316, 10.1113/jphysiol.2012.229690.
- Leena P. Bharath; Jae Min Cho; Seul-Ki Park; Ting Ruan; Youyou Li; Robert Mueller; Tyler Bean; Van Reese; Russel S. Richardson; Jinjin Cai; et al.Ashot SargsyanKarla PiresPon Velayutham Anandh BabuSihem BoudinaTimothy E. GrahamJ. David Symons Endothelial Cell Autophagy Maintains Shear Stress–Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arteriosclerosis, Thrombosis, and Vascular Biology 2017, 37, 1646-1656, 10.1161/atvbaha.117.309510.
- Anthony J. Donato; Ashley E. Walker; Katherine A. Magerko; R. Colton Bramwell; Alex D. Black; Grant D. Henson; Brooke R. Lawson; Lisa A. Lesniewski; Douglas R. Seals; Life-long caloric restriction reduces oxidative stress and preserves nitric oxide bioavailability and function in arteries of old mice. Aging Cell 2013, 12, 772-783, 10.1111/acel.12103.
- Christopher R. Martens; Douglas R. Seals; Practical alternatives to chronic caloric restriction for optimizing vascular function with ageing. The Journal of Physiology 2016, 594, 7177-7195, 10.1113/jp272348.
- Yun-Ying Wang; Ai-Fang Chen; Hua-Zhang Wang; Li-Yan Xie; Ke-Xu Sui; Qi-Yi Zhang; Association of shorter mean telomere length with large artery stiffness in patients with coronary heart disease. The Aging Male 2010, 14, 27-32, 10.3109/13685538.2010.529196.
- Pim van der Harst; Gerrit van der Steege; Rudolf A. de Boer; Adriaan A. Voors; Alistair Hall; Marcel J. Mulder; Wiek van Gilst; Dirk J. van Veldhuisen; Telomere Length of Circulating Leukocytes Is Decreased in Patients With Chronic Heart Failure. Journal of the American College of Cardiology 2007, 49, 1459-1464, 10.1016/j.jacc.2007.01.027.
- Athanase Benetos; Jeffrey P. Gardner; Mahmoud Zureik; Carlos Labat; Lu Xiaobin; Chris Adamopoulos; Mohamed Temmar; Kathryn E. Bean; Frédérique Thomas; Abraham Aviv; et al. Short Telomeres Are Associated With Increased Carotid Atherosclerosis in Hypertensive Subjects. Hypertension 2004, 43, 182-185, 10.1161/01.hyp.0000113081.42868.f4.
- Lisa D Moore; Thuc Le; Guoping Fan; DNA Methylation and Its Basic Function. Neuropsychopharmacology 2012, 38, 23-38, 10.1038/npp.2012.112.
- Samira Tabaei; Seyyedeh Samaneh Tabaee; DNA methylation abnormalities in atherosclerosis. Artificial Cells, Nanomedicine, and Biotechnology 2019, 47, 2031-2041, 10.1080/21691401.2019.1617724.
- Hui Xu; Shuang Li; You-Shuo Liu; Roles and Mechanisms of DNA Methylation in Vascular Aging and Related Diseases. Frontiers in Cell and Developmental Biology 2021, 9, 699374, 10.3389/fcell.2021.699374.
- Brian D Strahl; C. David Allis; The language of covalent histone modifications. Nature Cell Biology 2000, 403, 41-45, 10.1038/47412.
- Guo-Hua Ding; Dan-Di Guo; Yang Guan; Chun-Yu Chi; Bao-Dong Liu; Changes of DNA methylation of Isoetes sinensis under Pb and Cd stress. Environmental Science and Pollution Research 2018, 26, 3428-3435, 10.1007/s11356-018-3864-3.
- Marta Halasa; Kamila Adamczuk; Grzegorz Adamczuk; Syeda Afshan; Andrzej Stepulak; Marek Cybulski; Anna Wawruszak; Deacetylation of Transcription Factors in Carcinogenesis. International Journal of Molecular Sciences 2021, 22, 11810, 10.3390/ijms222111810.
- Munehiro Kitada; Yoshio Ogura; Daisuke Koya; The protective role of Sirt1 in vascular tissue: its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290-2307, 10.18632/aging.101068.
- Julia Beermann; Maria-Teresa Piccoli; Janika Viereck; Thomas Thum; Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiological Reviews 2016, 96, 1297-1325, 10.1152/physrev.00041.2015.
- Adam Harvey; Augusto C. Montezano; Rheure Alves Lopes; Francisco Rios; Rhian M. Touyz; Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Canadian Journal of Cardiology 2016, 32, 659-668, 10.1016/j.cjca.2016.02.070.
- Jason L. Johnson; Sarah J. George; Andrew C. Newby; Christopher L. Jackson; Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proceedings of the National Academy of Sciences 2005, 102, 15575-15580, 10.1073/pnas.0506201102.
- Ji Yoon Park; Jun Hyoung Park; Wookju Jang; In-Kwan Hwang; In Ja Kim; Hwa-Jung Kim; Kyung-Hyun Cho; Seung-Taek Lee; Apolipoprotein A-IV is a novel substrate for matrix metalloproteinases. The Journal of Biochemistry 2011, 151, 291-298, 10.1093/jb/mvr137.
- Jon M. Florence; Agnieszka Krupa; Laela M. Booshehri; Timothy C. Allen; Anna K. Kurdowska; Metalloproteinase-9 contributes to endothelial dysfunction in atherosclerosis via protease activated receptor-1. PLOS ONE 2017, 12, e0171427, 10.1371/journal.pone.0171427.

