Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sana Parveen | -- | 5771 | 2022-08-26 17:59:45 | | | |
| 2 | Amina Yu | -12 word(s) | 5759 | 2022-08-29 03:26:32 | | | | |
| 3 | Amina Yu | Meta information modification | 5759 | 2022-08-29 03:29:42 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Haque, N.; Parveen, S.; Tang, T.; Wei, J.; Huang, Z. Marine Bioactive Compounds Available on the Market. Encyclopedia. Available online: https://encyclopedia.pub/entry/26556 (accessed on 27 July 2026).
Haque N, Parveen S, Tang T, Wei J, Huang Z. Marine Bioactive Compounds Available on the Market. Encyclopedia. Available at: https://encyclopedia.pub/entry/26556. Accessed July 27, 2026.
Haque, Neshatul, Sana Parveen, Tingting Tang, Jiaen Wei, Zunnan Huang. "Marine Bioactive Compounds Available on the Market" Encyclopedia, https://encyclopedia.pub/entry/26556 (accessed July 27, 2026).
Haque, N., Parveen, S., Tang, T., Wei, J., & Huang, Z. (2022, August 26). Marine Bioactive Compounds Available on the Market. In Encyclopedia. https://encyclopedia.pub/entry/26556
Haque, Neshatul, et al. "Marine Bioactive Compounds Available on the Market." Encyclopedia. Web. 26 August, 2022.
Copy Citation
Marine natural products are potent and promising sources of drugs among other natural products of plant, animal, and microbial origin. Marine drugs are classified into six categories, where the basis of classification is nonuniform but maintains the flow of context within the category. Most of the drugs are categorized on the basis of the complexity of structures such as “spongonucleosides”, “antibody-drug conjugates”, and “peptides or proteins used as drugs or used in drug preparations”, but some are categorized on the basis of their mechanism of action, such as “microtubule inhibitors” and “deoxyribonucleic acid (DNA) alkylating agents”, or their natural source of abundance, such as “fish oil and its components as drugs”.
marine natural products
marine drugs
microtubule inhibitors
drug conjugated with an antibody
peptides or proteins
fish oil
1. Nucleoside Analogs
1.1. Cytarabine
Cytarabine, aka 1-β-d-Arabinofuranosylcytosine, Ara-C, CYTOSAR-U® (Pfizer, New York City, NY, USA), and DEPOCYT® (Pacira Pharma, San Diego, CA, USA; Bedford Lab, Seattle, DC, USA, Enzon Pharmaceuticals, Piscataway, NJ, USA), is the synthetic analog of naturally occurring spongothymidine. Cytarabine is among the most successful antitumor agents and is used even today, more than 50 years after its first approval in 1969 [1][2], for the treatment of acute leukemia [3]. Cytarabine is a prodrug and is phosphorylated intracellularly to produce the active molecule ara-cytidine-5′-triphosphate (ara-CTP). Intracellular conversion of ara-C to ara-CTP increases the bioactive concentration of ara-CTP before it acts rapidly as a potent inhibitor of mammalian DNA synthesis (Figure 1) [4][5][6][7]. The cytotoxic effect is mediated by inhibiting DNA polymerase, leading to megaloblastosis [5], as well as by the incorporation of ara-CTP in the growing DNA, resulting in a faulty DNA strand [8][9]. The faulty DNA eventually matures into aberrant chromosomes with multiple chromatid breaks and fragmentation in S phase of the cell cycle, which leads to cell death [10][11].
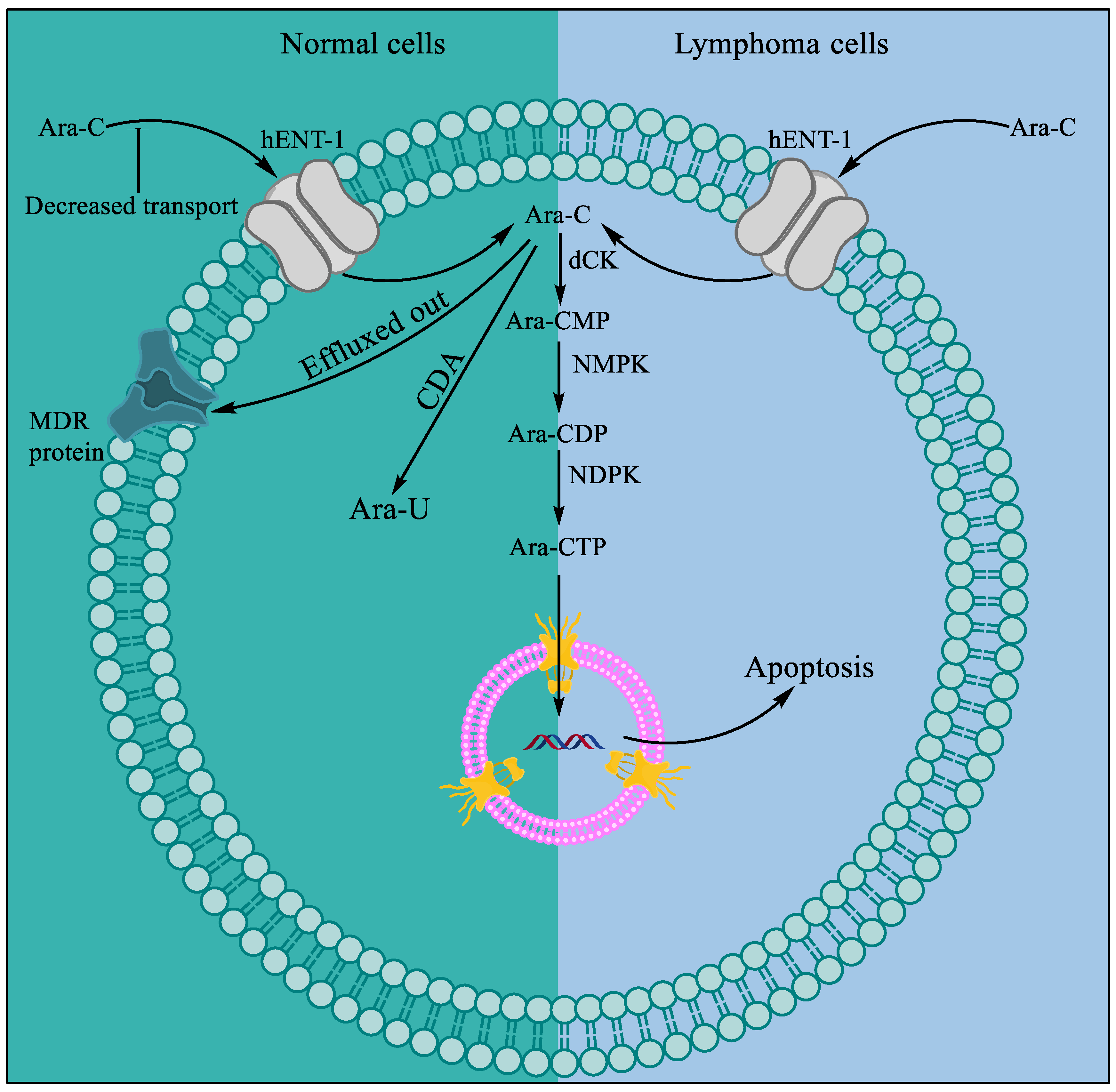
Figure 1. Mechanism of resistance to ara-C in normal cells and cytotoxicity in lymphoma. Compared to normal cells, lymphoma cells show more influx and less efflux of ara-C. A series of phosphorylation events by deoxycytidine kinase (dCK), followed by nucleoside monophosphate kinase (NMPK) and nucleoside diphosphate kinase (NDPK), occurs that convert ara-C to bioactive ara-CTP.
Ara-C is readily deaminated to its inactive form, uracil arabinoside, by cytidine deaminase (CDA) present in the intestine and liver, when administered orally [9][12]. Intravenous administration of ara-C produces comparatively higher active drug concentrations in plasma and cerebrospinal fluid, where 80% of the drug is excreted as uracil arabinoside in urine [13]. In 1999, DepoCyt® (Pacira Pharmaceuticals, San Diego, CA, USA), a liposomal formulation of cytarabine for sustained delivery of the drug, was approved by the US Food and Drug Administration (FDA) for lymphomatous meningitis treatment [14][15]. The liposomal formulation of cytarabine alone or in combination with anthracyclines such as daunorubicin is more effective because of slow and sustained delivery and possible evasion of nanosized liposomes from the drug efflux pump as they enter the cells intact [16][17][18]. The common adverse events observed in patients receiving cytarabine treatment include anorexia, nausea, vomiting, diarrhea, bleeding, and myelosuppression. Following prolonged intravenous or intrathecal administration of cytarabine, risks for central nervous system (CNS) toxicity and renal and hepatic failure have been reported [19][20].
1.2. Vidarabine
Vidarabine, aka 9-β-D-Arabinofuranosyladenine, Ara-A, and VIRA-A® (King Pharmaceuticals, Bristol, TN, USA), is a synthetic analog of arabinonucleosides inspired by naturally occurring spongothymidine and spongouridine. Vidarabine selectively inhibits DNA viruses [21][22] but exhibits little or no inhibition of RNA viruses [23]. Vidarabine was principally synthesized as a potential anticancer drug [24] but was formally approved by the US FDA in 1976 as an ophthalmic ointment (3%) with indications for the treatment of superficial keratosis resistant to idoxuridine caused by herpes simplex virus (HSV), keratoconjunctivitis, and recurrent epithelial keratitis caused by HSV1/2 [25]. However, the drug was discontinued from the market because of the availability of a better commercial alternative [26][27]. The therapeutically active concentration of the drug was not reached because of its relatively low solubility and high body clearance, and the drug is rapidly deaminated to its inactive form arabinosyl hypoxanthine by adenosine deaminase, restricting its clinical use to limited pathological conditions [7][28]. However, the drug was investigated on multiple occasions in combination with the adenosine deaminase inhibitor deoxycoformycin (dCF, pentostatin) but had little success [29][30]. Soon, attention was switched to the more potent alternative fludarabine (discussed in the next section) [7][24][31].
1.3. Fludarabine
Fludarabine, aka 9-β-d-Arabinofuranosyl-2-fluoroadenine-5’-phosphate, FLUDARA® (Sandoz, Basel, Switzerland) or OFORTA® (Sanofi-Aventis, Paris, France), is a synthetic purine nucleoside analog inspired by marine spongothymidine. The drug was initially approved for the treatment of adult B-cell chronic lymphocytic leukemia (CLL) in 1991 [32]. Fludarabine is more effective as a single agent for the treatment of CLL [32][33] than traditional chemotherapy using alkylating agents such as chlorambucil [34]. However, the administration of fludarabine in combination with other agents, such as cyclophosphamide, rituximab, mitoxantrone, and dexamethasone, has been proven to be a more effective treatment for CLL [35][36]. Fludarabine failed to produce therapeutic benefits in patients with other solid tumors because of the dose-limiting toxicity associated with myelosuppression [37][38][39][40]. Although fludarabine has a higher response rate than previous combination chemotherapeutic regimens, patients under treatment develop moderate to severe toxicity [32]. Severe indications of (a) bone marrow toxicity, such as thrombocytopenia and leukopenia [41][42], (b) pulmonary toxicity, such as cough, dyspnea, and hypoxia [43][44][45], and (c) neurotoxicity, such as visual field deficits, seizures, and encephalopathy [46][47], should be closely monitored in patients treated with a high dose of fludarabine to alleviate or mitigate the symptoms [32]. The mechanism of neurotoxicity remains elusive, but the evidence supports that, like adenosine, fludarabine crosses the blood-brain barrier and acts as an A1 receptor agonist, causing abnormal synaptic function [48][49]. On the other hand, pulmonary toxicity usually responds to steroids [32][50].
Fludarabine, a phosphorylated form of fluorinated arabinofuranosyl adenine, is resistant to deamination and more soluble than ara-A [51]. The drug is metabolized to a dephosphorylated form (F-ara-A) before being transported into the cell. The drug F-ara-A is administered orally, as well as parentally [7], and the clearance is mostly through urine, the degree of which depends on the dosage amount, duration, schedule, and patient metabolism [7][52][53]. F-ara-A is rephosphorylated to F-ara-adenosine-triphosphate (F-ara-ATP) by subsequent phosphorylation in the cell [54][55], where it accumulates to the bioactive concentration required for cytotoxicity by competing with cellular nucleotides and incorporating into the nucleic acid [56][57][58].
1.4. Nelarabine
Nelarabine, aka 2-Amino-9-α-d-arabinofuranosyl-6-methoxy-9H-purine, ARRANON® (GSK, Brentford, UK) ATRIANCE® (Novartis, Basel, Switzerland), is also a synthetic purine nucleoside analog inspired by marine spongothymidine. Nelarabine is a chemotherapeutic anticancer prodrug that is first metabolized in cells to ara-G followed by phosphorylation to the active form ara-guanosine triphosphate (ara-GTP). Ara-GTP competes with dGTP for DNA polymerase and is incorporated into the nucleic acid, causing DNA breakage and cell death [59][60]. Unlike the previously mentioned nucleoside analogs, nelarabine is more water-soluble [59][60]. The half-life of the drug is 30 min, and it is mostly excreted out with urine. Inspired by the cytotoxic properties of cytarabine and fludarabine, nelarabine was tested against various hematological malignancies and was approved by the FDA in 2005 [61] due to its selective cytotoxic activity against T-cell acute lymphoblastic leukemia (ALL) or T-cell lymphoblastic lymphoma (ABL). Another rationale for investigating ara-G analogs as treatments for T-cell malignancies is based on the report that patients with purine nucleoside phosphorylase deficiency, an autosomal-recessive rare disease, suffer from T-cell lymphopenia-related immunodeficiency. This observation led to the hypothesis that the water-soluble purine analog might exert selective cytotoxic effects on T-cell-related malignancies [61][62][63]. The most likely cause of the T-cell toxicity of nelarabine is the higher concentration of ara-G in T cells than in B cells [62][63].
Nonhematological adverse events associated with nelarabine treatment are mostly mild to moderate and are either reversible or can be alleviated by the appropriate medications. Neurological adverse events that may be severe mainly limit the use of nelarabine for ALL and ABL. Patients administered nelarabine treatment must be kept under close observation, and the treatment should be stopped at any sign of neurological adverse events [63][64][65].
1.5. Histochrome®: Sodium Salt of Echinochrome A—A Common Sea Urchin Pigment
Histochrome® from Pacific-Ocean Institute of Bioorganic Chemistry (Vladivostok, Russia) (Figure 2) is another secondary metabolite from the marine organism Scaphechinus mirabilis (sea urchin), which has been in clinical use since 1999 [66][67]. The drug exerts a therapeutic cytoprotective effect and is used mostly in Russia to treat a variety of diseases, such as degeneration of the macula, retina, and cornea, circulatory disorder of the retina, and myocardial ischemia/reperfusion injury [68]. The cytoprotective action of the drug is derived from its ability to protect against DNA damage by regulating the apoptosis cascade [67].
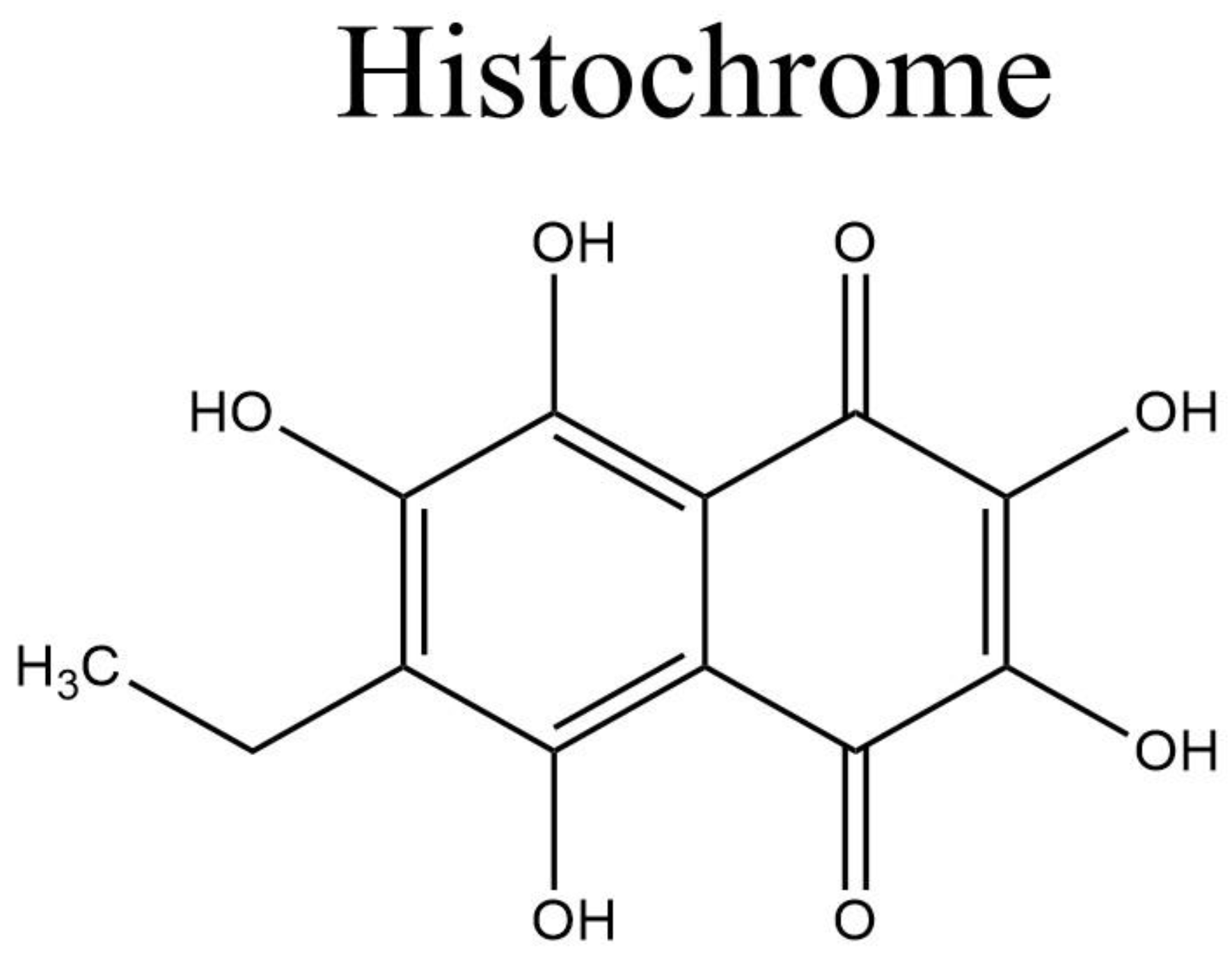
Figure 2. Histochrome structure.
2. Microtubule Inhibitors
Eribulin mesylate or EM, aka E7389 and HALAVEN® (Eisai, Bunkyo, Japan) (Figure 3B) is a synthetic analog of halichondrin B (Figure 3A), a polyether macrolide discovered in the marine sponge Halichondria okadai [69]. Similar to halichondrin B, EM also inhibits tumor cell proliferation by arresting cells in G2–M phase of the cell cycle [69][70][71]. EM was approved as a treatment for metastatic breast cancer in patients with a previous history of chemotherapeutic treatment in 2010 by the US FDA [72] and in 2011 by Health Canada [73]. In the following year (2016), EM was also approved for the treatment of metastatic liposarcoma [71][74][75].
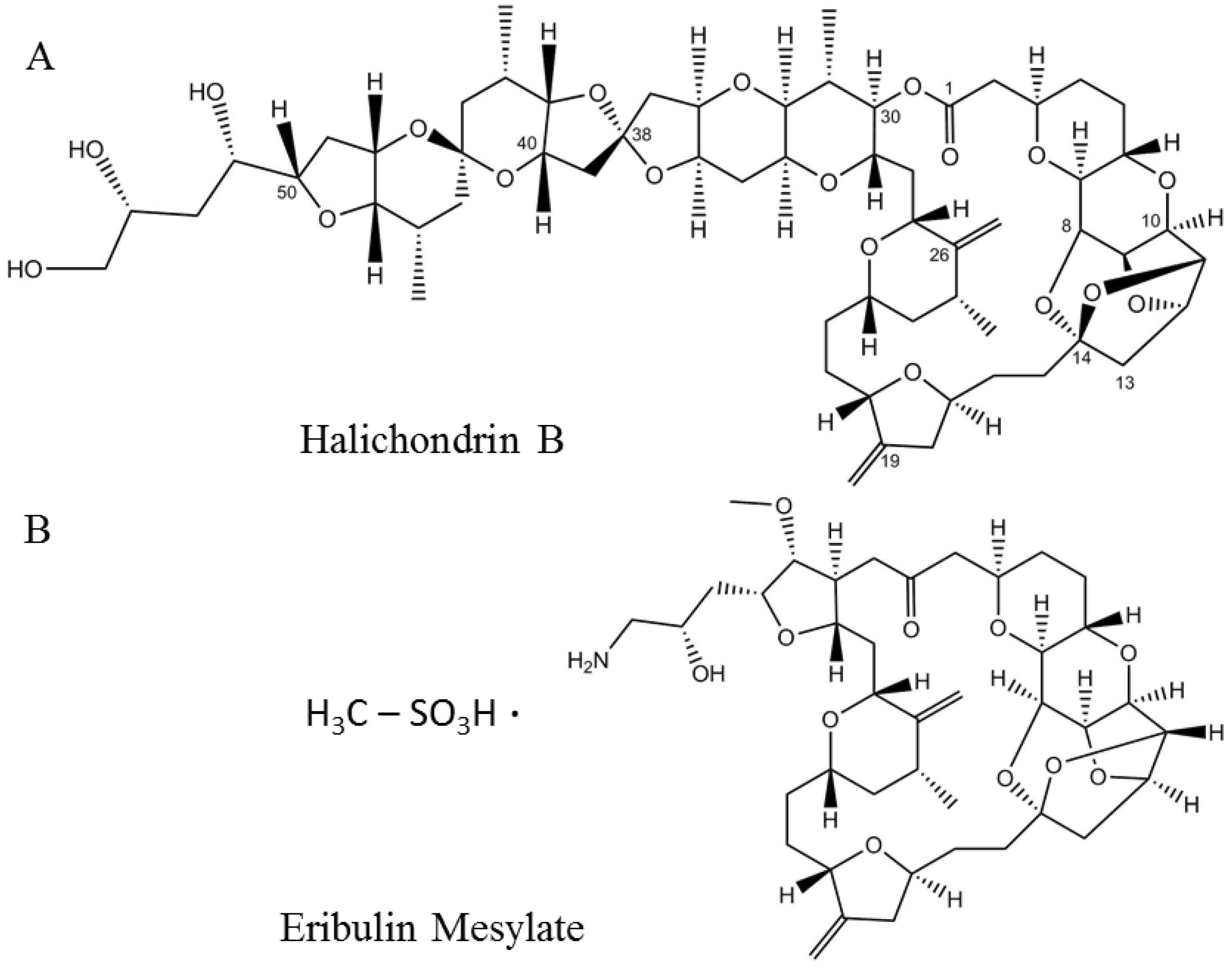
Figure 3. Structures of the naturally occurring marine compound (A) halichondrin B and its synthetic analog (B) eribulin mesylate.
Halichondrin B belongs to the family of halichondrin natural compounds. Members of this chemical class are categorized into halichondrin, norhalichondrin, and homohalichondrin [69]. Halichondrin B is a more potent inhibitor than most of the known microtubule inhibitors and works by producing nonproductive aggregates of tubulin [70]. The rare abundance of the organism and tedious process of the total synthesis of such a large polyether macrolide (1111.329 Da) encouraged the discovery of a minimum pharmacophore analog by the Kishi group in 2001 [76], which was named ER-086526 (later renamed eribulin, E7389). EM was shown to inhibit the proliferation of multiple cell lines at subnanomolar concentrations through a mechanism consistent with the parent molecule halichondrin B [76].
3. DNA Alkyating Agents
3.1. Trabectedin
Trabectedin, aka ET-743 and YONDELIS® (PharmaMar SA, Madrid, Spain), is a synthetic antineoplastic alkylating agent originally extracted from the tunicate Ecteinascidia turbinate [77]. The molecule consists of three tetrahydroisoquinoline alkaloid moieties interconnected with each other in a nearly rigid and complex structure (Figure 4A) [78]. The natural abundance of the drug, which is as low as 1 g per 1000 kg of tunicate, does not meet the need for preclinical and clinical experiments, which forced chemists to find a route for its chemical synthesis. Total synthesis of trabectedin and lurbinectedin was accomplished in 26 individual steps with a yield of 1.6% [79][80]. The drug was designated as an orphan [81] and approved by the European Medicines Agency (EMA) in 2007 for the treatment of soft-tissue sarcoma [82][83] and in combination with pegylated liposomal doxorubicin (PLD) for the treatment of relapsed platinum-sensitive ovarian cancer [84][85]. Yondelis® (PharmaMar SA, Madrid, Spain) was further approved by the US FDA in 2015 for the treatment of liposarcoma or leiomyosarcoma that is unresectable or has metastasized [86]. Several adverse events, which are classified as moderate to high but manageable, have been reported during clinical trials of trabectedin, such as neutropenic sepsis, rhabdomyolysis, hepatotoxicity, cardiomyopathy, capillary leak syndrome, and embryo-fetal toxicity, which should be monitored during treatment [86][87]. The drug gets metabolized to many different substituted compounds and is mostly excreted out in feces and urine; the half-life is reported to be about 26 h [88][89].
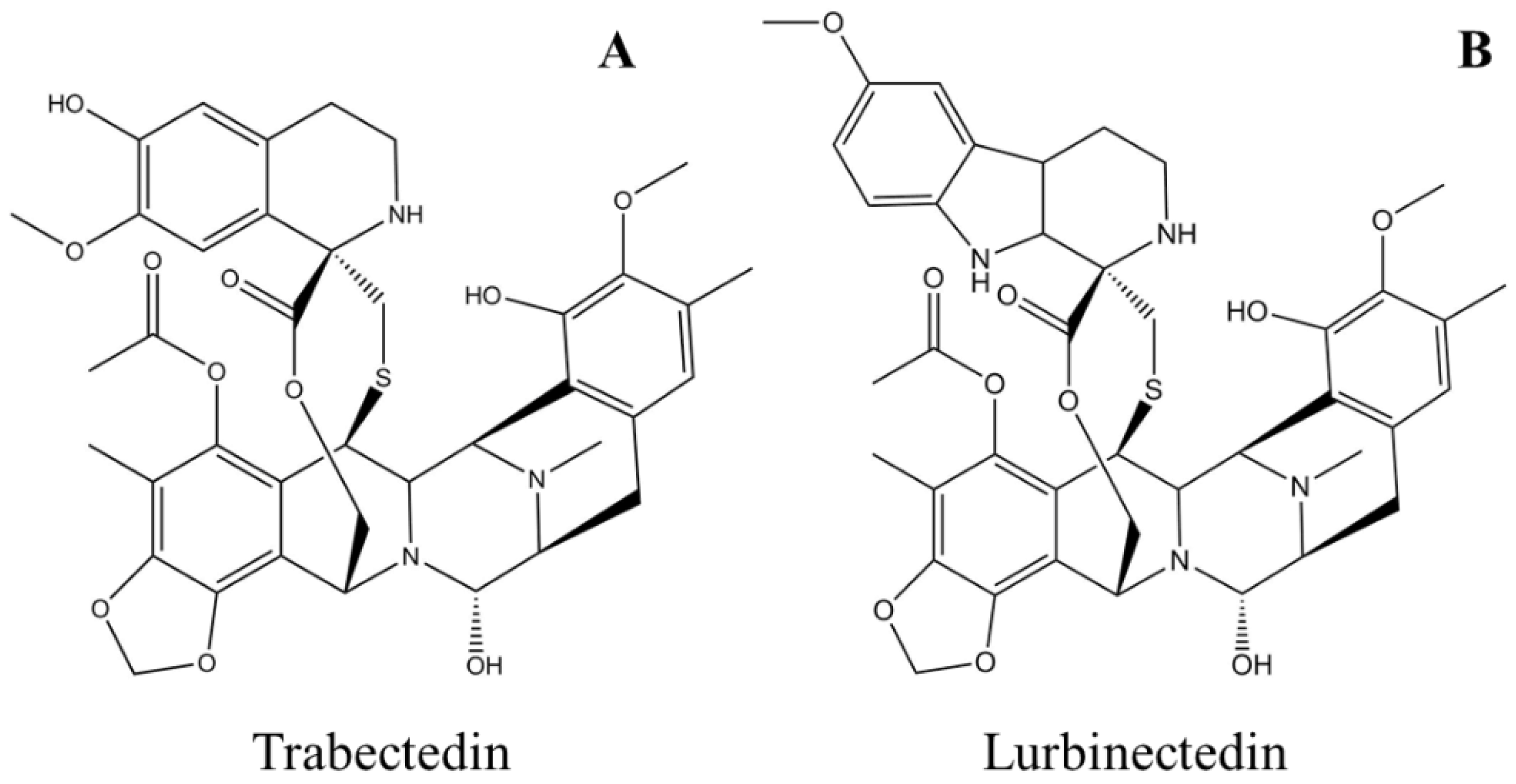
Figure 4. Schematic molecular representations of (A) trabectedin and (B) lurbinectedin.
Unlike most DNA alkylating agents, trabectedin binds to the minor groove guanine at the N2 position, initiating a cascade of events inducing cancer cell apoptosis. Trabectedin binding bends the DNA toward the major groove with drug protrusion on the convex side, and DNA subsequently loses affinity for its binding proteins, generating double-strand breaks [90][91][92] (Figure 5A). Specifically, trabectedin blocks cells in the G2–M phase of the cell cycle [93]. In addition to the general mechanism of cytotoxicity, trabectedin exerts atypical cell type-specific cytotoxicity. Cells deficient in nucleotide excision repair (NER) are 2–10 times less sensitive to trabectedin [94], whereas cells deficient in homologous repair (HR) are approximately 100 times more sensitive; however, in cells with nonhomologous end-joining (NHEJ) repair, no such differences were observed [95]. Trabectedin shows an atypical mechanism of the inhibition of tumor cell proliferation by interfering with the DNA repair machinery and inhibiting the active transcription process (Figure 5A).
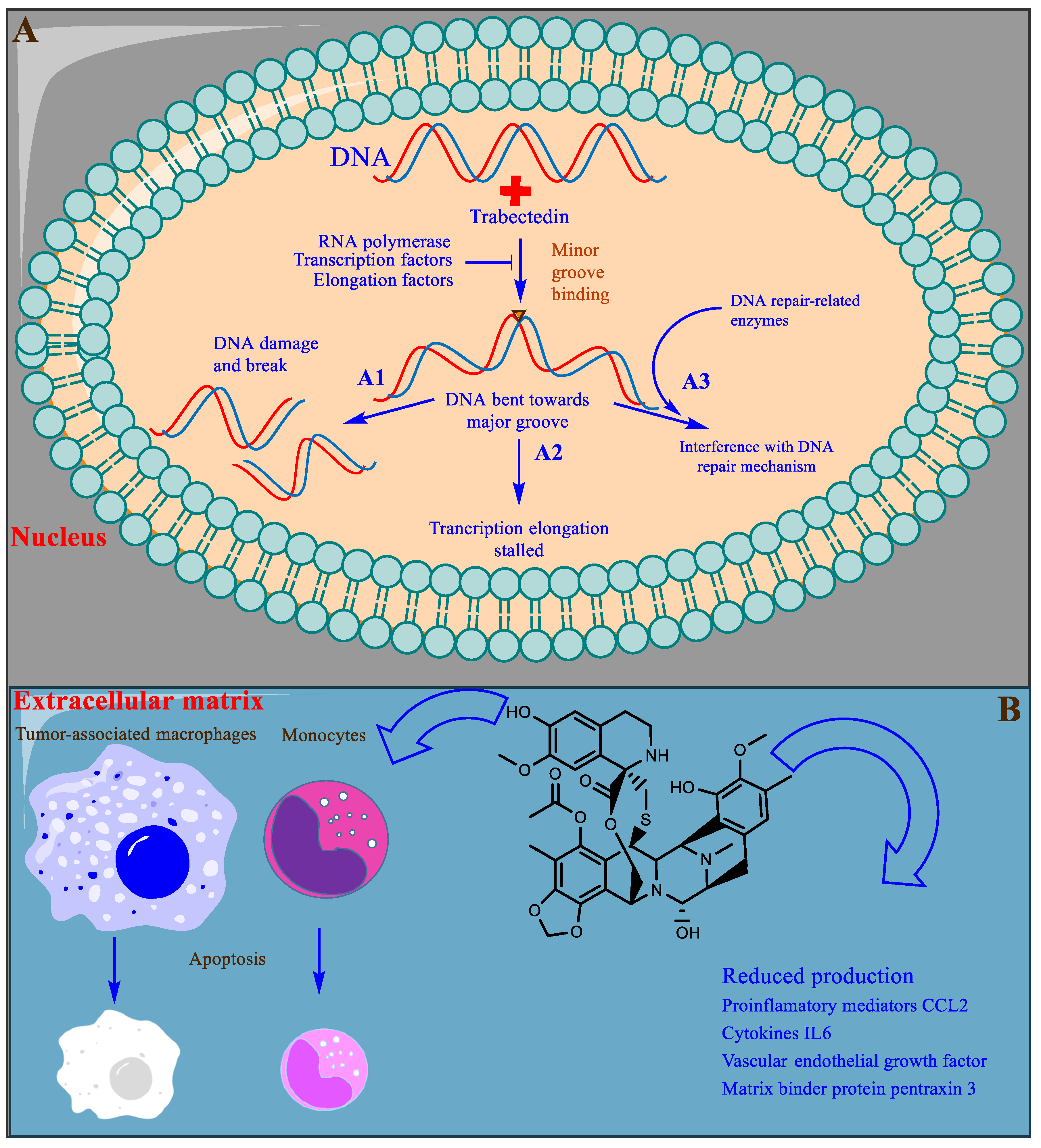
Figure 5. A representative illustration of the mechanism of trabectedin action. (A) The bilayer shows the nucleus, DNA strands are shown in red and blue stranded, and the inverted triangle in the minor groove represents trabectedin binding. (A1) DNA damage caused by trabectedin. (A2) The inhibition of transcription elongation by the inhibition of transcription factors and other related enzymes. (A3) The interference of trabectedin with the DNA repair machinery. (B) The extracellular matrix and the tumor microenvironment. TAMs and monocytes undergo cell death following inhibition by trabectedin, and trabectedin is shown in 2D in black stick representation.
Partial or indirect antitumor activity is also escalated by trabectedin following the disruption of the protumor functions of tumor-associated macrophages (TAMs) and monocytes present in the tumor microenvironment. TAMs produce many growth factors (GFs) (epidermal GF, fibroblast GF, and vascular endothelial GF), extracellular matrix-degrading enzymes, and immunosuppressive cytokines that are required for tumor proliferation and evasion of detection by the immune system [96]. Trabectedin shows selective cytotoxicity toward TAMs and monocytes, indirectly inhibiting tumor growth (Figure 5B) [97]. Trabectedin is unique among chemotherapeutic agents because it has multitargeted antineoplastic properties. It is also proposed to possess enormous therapeutic potential and is currently under experimental investigation [98].
3.2. Lurbinectedin
Lurbinectedin, aka ZEPZELCA™ (PharmaMar SA, Madrid, Spain) is a synthetic analog of trabectedin in which the tetrahydroisoquinoline ring is replaced with tetrahydro β-carboline (Figure 4B) [99]. Lurbinectedin was granted orphan status and was approved for the treatment of adult metastatic small-cell lung cancer (SCLC) in 2020 by the US FDA [100][101]. Important adverse events observed in clinical trials were myelosuppression, hepatotoxicity, and embryo-fetal toxicity. Appropriate monitoring protocols should be implemented before and during drug administration, such as blood count and liver function tests [102][103]. The drug half-life (51 h) in the body increases when administered with CYP450 3A inhibitors, and the drug gets excreted from the body mostly in feces and urine [100][104].
The results of a clinical investigation of the effects of lurbinectedin on various tumors, such as SCLC, ovarian cancer, and breast cancer, indicated better pharmacokinetic profiles, which may allow the administration of higher therapeutic concentrations with less toxicity [105][106][107][108][109]. Lurbinectedin acts in a similar manner to trabectedin by inducing alkylation-dependent DNA damage, inhibition of active transcription, modulation of the DNA repair pathway, and modulation of the tumor microenvironment; however, monocytes are more sensitive to lurbinectedin [107][110]. The selective inhibition of RNA polymerase II transcription in SCLC has proven very effective [107], but another serious challenge is to counter the loss-of-function driver mutations in the tumor suppressor genes RB1 and TP53 and the amplification of the MYC gene. Targeted therapy and immunotherapy are anticipated to prolong the progression-free survival of patients with SCLC [111].
4. Antibody-Drug Conjugates
4.1. Brentuximab Vedotin
Brentuximab Vedotin or BV, aka ADCERTIS® (Seattle Genetics, Bossel, USA) is an anti-CD30 antibody-containing ADC approved by the US FDA in 2011 for the treatment of various T-cell lymphomas. BV has been indicated for the treatment of adult patients with relapsed classic Hodgkin lymphoma (cHL), systemic anaplastic large-cell lymphoma (sALCL), primary cutaneous anaplastic large-cell lymphoma (pcALCL), or CD30 expressing mycosis fungoides (MF) [112].
CD30 is a cell surface glycoprotein that belongs to the tumor necrosis factor receptor superfamily (TNFRSF8, member 8). CD30 expression on normal cells is limited to a small population of activated B and T lymphocytes, natural killer cells, and some viruses (such as human immunodeficiency virus (HIV), T-lymphotropic virus-1 (HTLV-1), or Epstein-Barr virus (EBV))-infected cells. CD30 is also characteristically expressed in hematopoietic malignant cells in cancers such as cHL, sALCL, MF, and pcALCL [113][114]. The differential expression of CD30 on normal and hematopoietic malignant cells has directed researchers to explore the mechanism of action and its use in targeted cancer therapy. Chemotherapy, radiation therapy, and anti-CD30 antibody (alone/naked or conjugated with immunotoxin therapy) have a lower success rate in clinical trials than BV, which substantially prolongs progression-free survival [115].
Patients receiving BV require continuous observation, as various but mostly manageable toxicities have been observed, including peripheral neuropathy, anaphylaxis and infusion reactions, hematological toxicities, opportunistic infections, tumor lysis syndrome, hepatotoxicity, pulmonary toxicity, serious dermatological reactions, gastrointestinal complications, and embryo-fetal toxicity [112]. Freely circulating extracellular CD30-positive vesicles carrying ADCs may bind to CD30L-expressing cells, resulting in their death, which is probably the cause of off-target binding and toxicity (an in vitro study) [116]. BV has delivered better performance than the classical treatment regimen, but neuropathy and other adverse events may limit the long-term use of BV, which must be overcome by obtaining a better understanding of the role of CD30 in antiapoptotic mechanisms [114].
4.2. Polatuzumab Vedotin
Polatuzumab Vedotin or PV, aka POLIVY™ (Genentech, San Francisco, CA, USA) is an anti-CD79b antibody-containing ADC approved by the US FDA and EMA in 2019 [117]. PV administered in combination with bendamustine and a rituximab product (BR) is used for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). CD79b is expressed on most mature B cells; however, it is ubiquitously expressed on DLBCL cells, making it a logical target for disease treatment [118]. Other lymphomas, such as mantle cell lymphoma, Burkitt lymphoma, and follicular lymphoma (FL), have also been shown to express CD79b [119]. Among all non-Hodgkin lymphomas, 31% are DLBCL, and 50–70% are cured with BR treatment, but the disease relapses in a fraction of treated patients and has a dismal prognosis [120]. Similar to BV, PV also releases MMAE to cells expressing CD79b, a cell surface glycoprotein specific for PV [118]. Compared to the classical treatment regimen with BR, PV in combination with BR produces a twofold higher rate of complete remission, which formed the basis of the approval of the latter treatment regimen [121]. Although treatment with PV promises better and longer, healthier lives, PV is associated with toxicity similar to that of BV and should be used with extreme care [118][122].
4.3. Enfortumab Vedotin
Enfortumab vedotin or EV, aka Padcev® (Astellas Pharma US, Inc., Northbrook, USA) is an anti-necrin-4 antibody-containing ADC approved by the FDA in 2019 for the treatment of advanced or metastatic urothelial cancer [123]. EV is attached to MMAE via a protease-cleavable linker [124]. It also contains a fully human monoclonal antibody directed against Nectin-4, an extracellular adhesion protein that is expressed at high levels in urothelial cancers [125]. Similar to PV, after the enfortumab–Nectin-4 complex is internalized into the cell, EV releases MMAE to cells expressing Nectin-4 through proteolytic cleavage [126]. Subsequently, the microtubule network within the cell is disrupted, arresting the cell cycle and inducing apoptosis [124]. EV is an anticancer agent that destroys tumor cells by inhibiting their ability to replicate but not in patients with a moderate to severe hepatic impairment [124]. In addition, EV may also cause significant hyperglycemia, leading, in some cases, to diabetic ketoacidosis, and it should not be administered to patients with a blood glucose level >250 mg/dL [124].
4.4. Belantamab Mafodotin
Belantamab mafodotin or BM, aka Blenrep® (GlaxoSmithKline, Brentford, UK) is an anti-B-cell maturation antigen (BCMA) antibody-containing ADC approved by the FDA in 2020 [127]. BM has been indicated for the treatment of adults with relapsed or refractory multiple myeloma who have received at least four prior therapies, including an anti-CD38 monoclonal antibody, a proteasome inhibitor, and an immunomodulatory agent [128]. It treats patients with the aforementioned characteristics through antibody-dependent cell-mediated cytotoxicity, G2/M cell-cycle arrest, and apoptosis [129]. BM is an afucosylated monoclonal antibody that targets BCMA, which is uniquely expressed on CD138-positive myeloma cells and conjugated to the microtubule disrupter monomethyl auristatin-F (MMAF) [130]. Its afucosylation of the Fc region of BM enhances antibody-dependent cell-mediated cytotoxicity [129][130]. Because of its risk of adverse effects and long duration of action, as it is administered every 3 weeks, BM shows a narrow therapeutic index [128]. Extreme care is essential due to the high risk of keratopathy that has been reported during clinical trials of BM, with a rate of approximately 71% [128].
5. Peptides or Proteins Used as Drugs or in Drug Preparations
5.1. Plitidepsin
Plitidepsin, aka APLIDIN® (PharmaMar SA, Madrid, Spain) is a synthetic cyclic peptide that was originally extracted from the ascidian Aplidium albicans (sea squirt). Plitidepsin belongs to a class of compounds known as didemnins (Figure 6A) [131]. Plitidepsin is an analog of didemnin B (Figure 6B,C). Other marine tunicates, such as Trididemnum solidum, are also abundant sources of similar depsipeptides, such as didemnins A–E (Figure 6B), which also possess antitumor and antiviral activities [132][133]. Plitidepsin was approved for the treatment of relapsed and refractory multiple myeloma in combination with dexamethasone by the Therapeutic Goods Administration (TGA), Australia, in 2018 [134]. The drug was granted orphan status by the EMA in 2003 as a treatment for leukemia, but the EMA did not approve the drug for relapsed and refractory multiple myeloma in combination with dexamethasone, citing that “the drug risk did not outweigh its benefits” [135]. Following the EMA refusal for marketing authorization of Aplidin, a lawsuit was filed by PharmaMar SA; the EU general court annulled the EMA decision, and the application was extended for review in October 2020 [135]. Recently, in preclinical studies, Aplidin was shown to be approximately 28 times more efficient than remdesivir against coronavirus disease 2019 (COVID-19) [136].
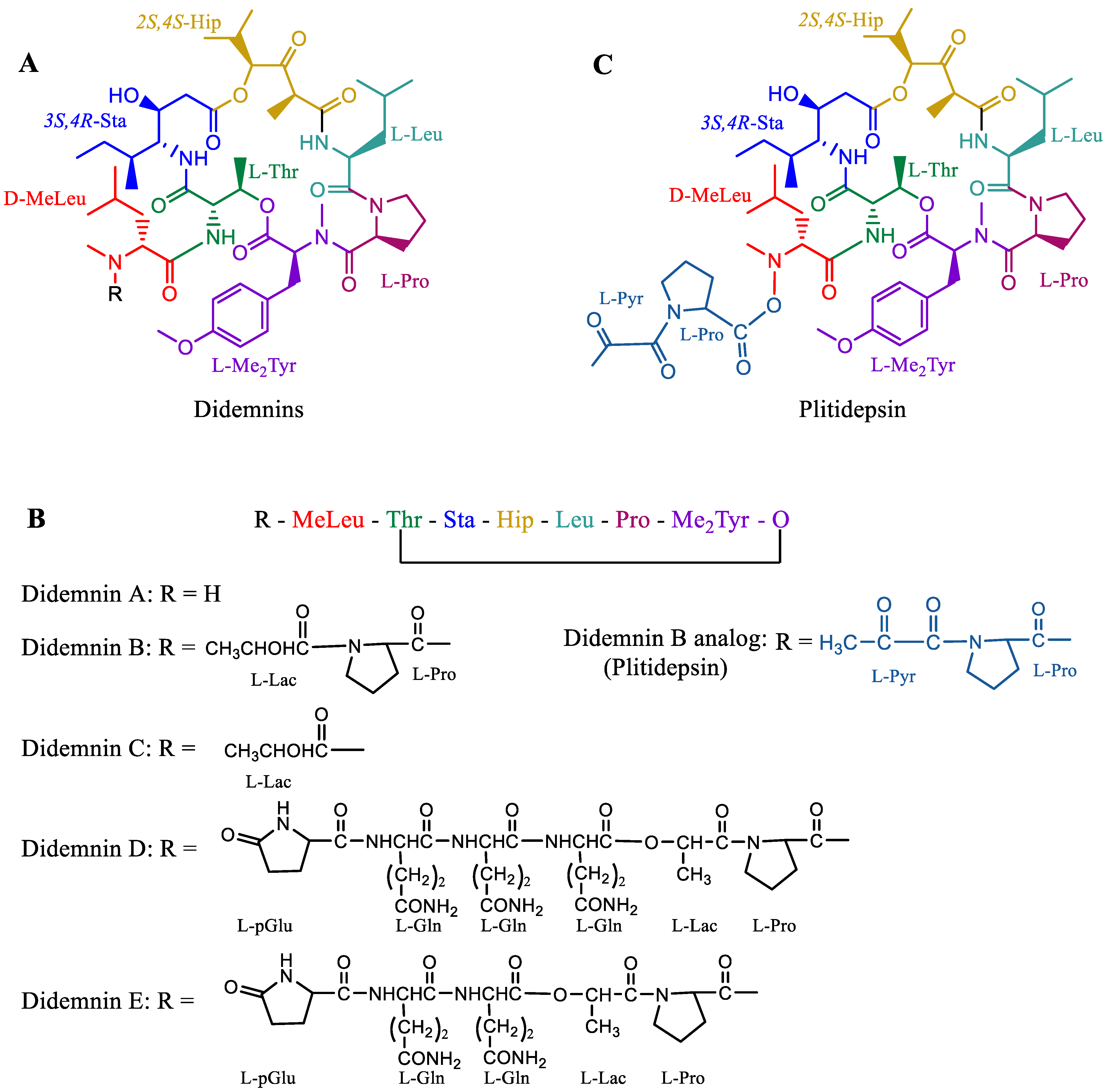
Figure 6. Molecules of the didemnin class. (A) General structure of didemnin, where “R” represents different substitutions in various didemnin members. (B) Simplified linear representation of a general didemnin. (C) Plitidepsin is similar to didemnin B, with a difference only in the terminal lactate that is oxidized to pyruvate. Short representations of residues: MeLeu—methyl leucine, Thr—threonine, Sta—statin, Hip—hydroxyisovalerylpropionic acid, Leu—leucine, Pro—proline, Me2Tyr—dimethyl tyrosine, Lac—lactate, Pyr—pyruvate, p-Glu—cyclic glutamate, and Gln—glutamine.
A phase III trial was conducted on the basis of the successful results from the phase II trial of Aplidin in patients with relapsed and refractory multiple myeloma. A significant improvement in the OS of the patients was reported for the drug in combination with dexamethasone (median OS of 11.6 months) compared to dexamethasone treatment alone (median OS 6.7 months) [137][138]. Most of the plitidepsin-related adverse events were manageable and well tolerated. Common adverse events reported were nausea, fatigue, and myalgia. Severe adverse events were related to hematological abnormalities such as anemia and lymphopenia that required continuous observation [139].
Multiple myeloma cells are often known to produce large amounts of misfolded proteins, which deplete free amino acids for new protein synthesis. The deposition of misfolded proteins is toxic to myeloma cells. These cancer cells devise mechanisms to destroy and recycle these misfolded proteins to obtain free amino acids and produce new proteins (Figure 7). Cereblon identifies misfolded proteins and redirects them for proteasomal degradation. The eEF1A2 protein, which is often overexpressed in myeloma cells, binds to misfolded proteins and redirects them for proteasomal degradation. When proteasomes do not function well or are inhibited, eEF1A2 forms a cluster of misfolded proteins after binding to them, which are subsequently destroyed by the aggresome during autophagy. Plitidepsin binds to eEF1A2 and renders it inefficient for binding with misfolded proteins and, hence, inhibits the destruction of misfolded proteins and cell apoptosis [140][141]. The tradeoff between the therapeutic efficiency and toxicity of the drug has remained controversial for a decade, which has kept researchers’ opinions split. However, the unique mechanism of the drug is promising for the treatment of multiple myeloma. Researchers have yet to discover appropriate combinations of drugs that may pave the way for the global clinical use of Aplidin.
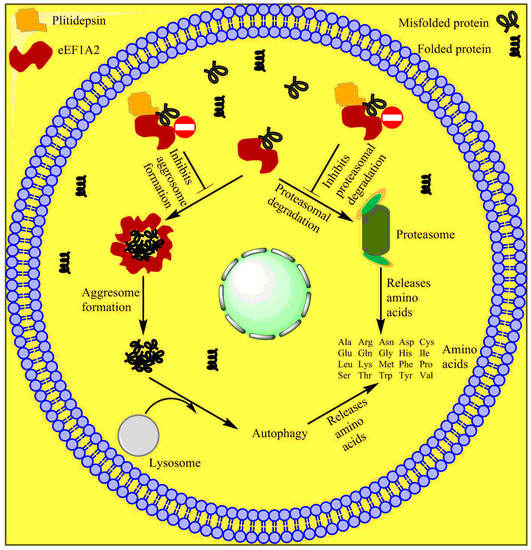
Figure 7. Mechanism of action of plitidepsin. Plitidepsin binds with eEF1A2 and blocks proteasomal aggresome degradation of misfolded proteins.
5.2. Ziconotide
Ziconotide, aka PRIALT®, Azur Pharma International Limited, Dublin, Ireland) (Figure 8A,B) is a disulfide-rich and 25 amino-acid long conotoxin peptide (or conopeptide) that was originally extracted from the venom (i.e., a cocktail of toxic peptides) of the marine cone snail Conus magus and is used by the organism to hunt its prey and defend against predators [142]. More than 750 species of the genus Conus are known to produce 100s of toxic peptides in their venom, accounting for ~10,000 different conopeptides [143]. These toxins have attracted increasing interest among drug developers because of their magnificent potency and selectivity toward mammalian ion channels, G-protein-coupled receptors, transporters, and enzymes. Conotoxins are 10–40 amino acids long, mostly disulfide-rich (two to three disulfide bonds) and are classified on the basis of their sequence, disulfide framework or type of mammalian target they bind [144].
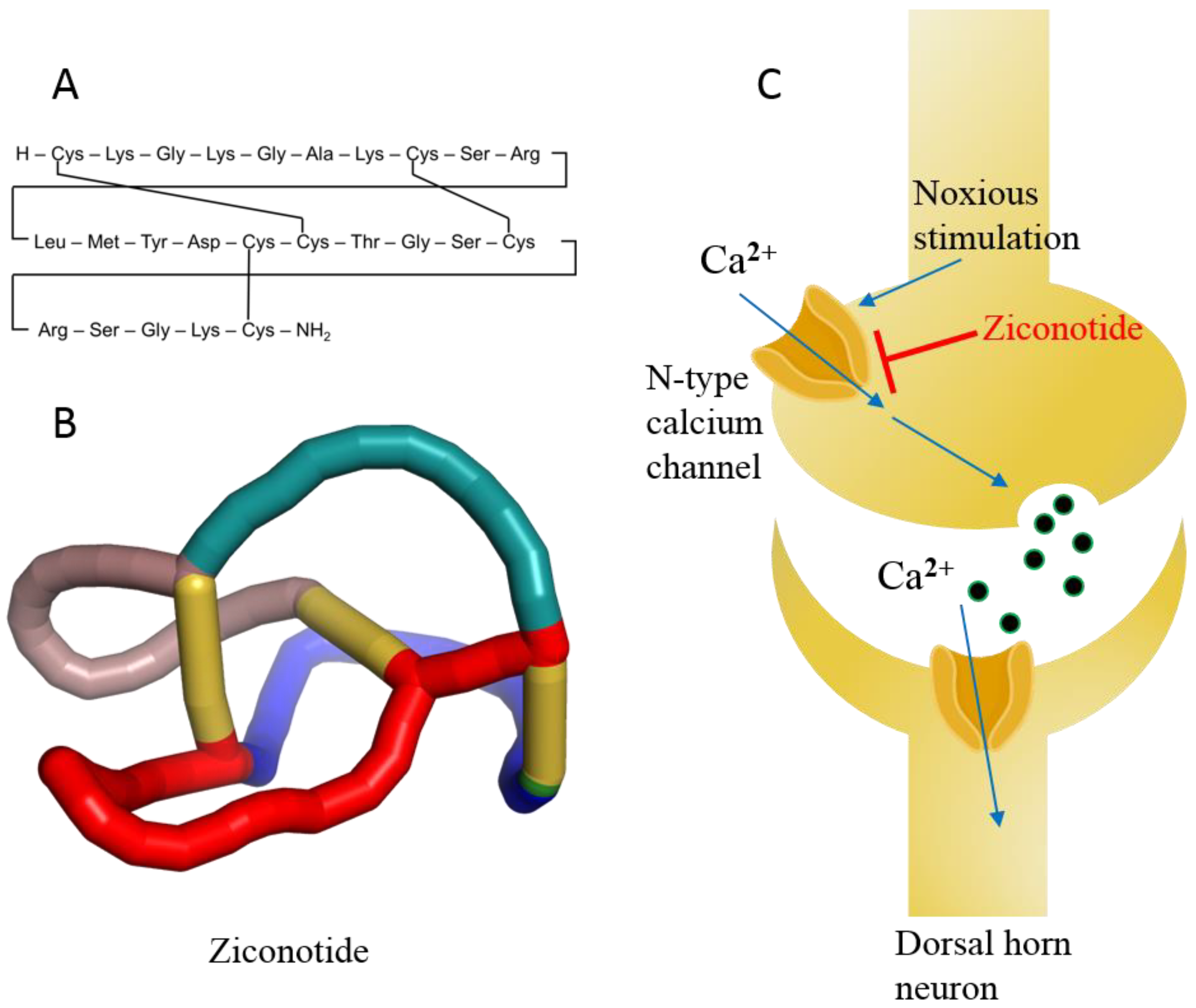
Figure 8. (A) Linear model of ziconotide with cross-linking of disulfide bonds. (B) A 3D knotted model of ziconotide. (C) Schematic diagram of ziconotide blocking the N-type calcium channel.
Ziconotide is the synthetic analog of ω-conotoxin and is highly soluble in water. In addition, ziconotide is the only nonopioid drug approved for the treatment of severe chronic neuropathic pain [145]. The drug is nonaddictive and is over 1000 times more effective than the classical opioid drug morphine, which is used for chronic pain. Ziconotide was approved in 2004 by the US FDA and in 2005 by the EMA [146]. Ziconotide is the only conotoxin approved for clinical use after three decades of research on the 1000s of conotoxins.
Ziconotide is an N-type voltage-gated ion channel blocker that inhibits the release of pro-nociceptive neurotransmitters (norepinephrine) and neuropeptides, relieving pain (Figure 8C) [146][147]. The median half-life of ziconotide in cerebrospinal fluid is approximately 4.5 h upon intrathecal injection, and it is completely cleared in 24 h. The metabolism of ziconotide is by endo- and exo-peptidases [148][149]. The most common adverse events reported during clinical trials are dizziness, nausea, nystagmus, confusion, gait abnormalities, memory impairment, blurred vision, headache, asthenia, vomiting, and somnolence [150]. Most of the adverse events associated with ziconotide monotherapy are mild to moderate and disappear over time [146][147][150]. The treatment of patients with chronic pain was also discontinued in some cases [151]. Therefore, experts believe that the psychological state of patients must be assessed a priori before treatment with ziconotide [152]. Ziconotide has been in clinical use for more than a decade and holds precedence for cancer-related and some noncancer-related pain management because of its high benefit-to-risk ratio compared to other opioid drugs, even today [153][154]. Experts also propose that this class of diverse peptide drugs may contribute to the list of therapeutic medicines for various diseases in the future because of its selectivity toward receptors, ion channels, and enzymes [145].
5.3. Protamine Sulfate
Protamine sulfate is the oldest drug of marine origin approved by the US FDA and has been in clinical use since 1939 [155][156] for heparin overdose. Marketing authorization of protamine sulfate is held by many pharmaceutical companies under various trade names, such as Prosulf® (CP Pharm and Wockhardt, Mumbai, India, Wales, UK) and Protam® (Eipico, Ramadan City, Egypt) [157]. Protamine sulfate is a strongly basic peptide containing more than two-thirds of the amino acid arginine. Protamine was originally obtained from the salmon sperm head, where it remained bound to negatively charged nucleic acids [158]. Protamine is 33 amino acids long and possesses the sequence MET PRO ARG ARG ARG ARG SER SER ARG PRO VAL ARG ARG ARG ARG ARG PRO ARG VAL SER ARG ARG ARG ARG ARG ARG GLY GLY ARG ARG ARG ARG (UniProt ID P69014). Because of its unique sequence, protamine may possess a random coil structure in solution or develop a secondary structure when bound to DNA. Positively charged arginine is present in four clusters, which prevents it from attaining a specific secondary structure because of strong charge repulsion [159]. Protamine is used as an antidote for heparin-induced anticoagulation in patients undergoing hemodialysis and heart surgery. Heparin neutralization usually occurs within 5 min of an intravenous protamine injection and is predominantly cleared from the body through urine [160].
Blood coagulates at the site of injury through the activation of many coagulation factors and serine proteases, such as IXa, Xa, Xia, and XIIa, resulting in the conversion of prothrombin to thrombin that finally converts fibrinogen into fibrin, which subsequently forms a mesh-like structure (called a clot) [161]. Low-molecular-weight heparin (LMWH) binds antithrombin III (a serine protease inhibitor) and enhances its affinity for factor Xa, preventing the formation of thrombin, which ultimately inhibits fibrin formation at the site of injury [162]. Cationic protamine binds to anionic LMWH, rendering antithrombin III free and resulting in an intrinsic coagulation process (Figure 9) [163]. Protamine remains successful in removing heparin from plasma, but albumin-bound heparin is released over time and restores the anticoagulant effect. A high dose of protamine is often associated with adverse events in patients who have been administered heparin during heart surgery. Common adverse events associated with protamine use are allergic reactions, low blood pressure, dyspnea, nausea, vomiting, lassitude, and back pain, among others [157]. Although protamine has a satisfactory performance in many patients undergoing heart surgery, it is often associated with severe adverse events, which has reduced its market value. Active research is underway to identify less toxic and noninvasive alternative agents for anticoagulation reversal in addition to the approved drug Andexxa® (Portola Pharmaceutical, South San Francisco, USA in 2018) [164]. Many more protamine alternatives are expected to be developed in the near future.
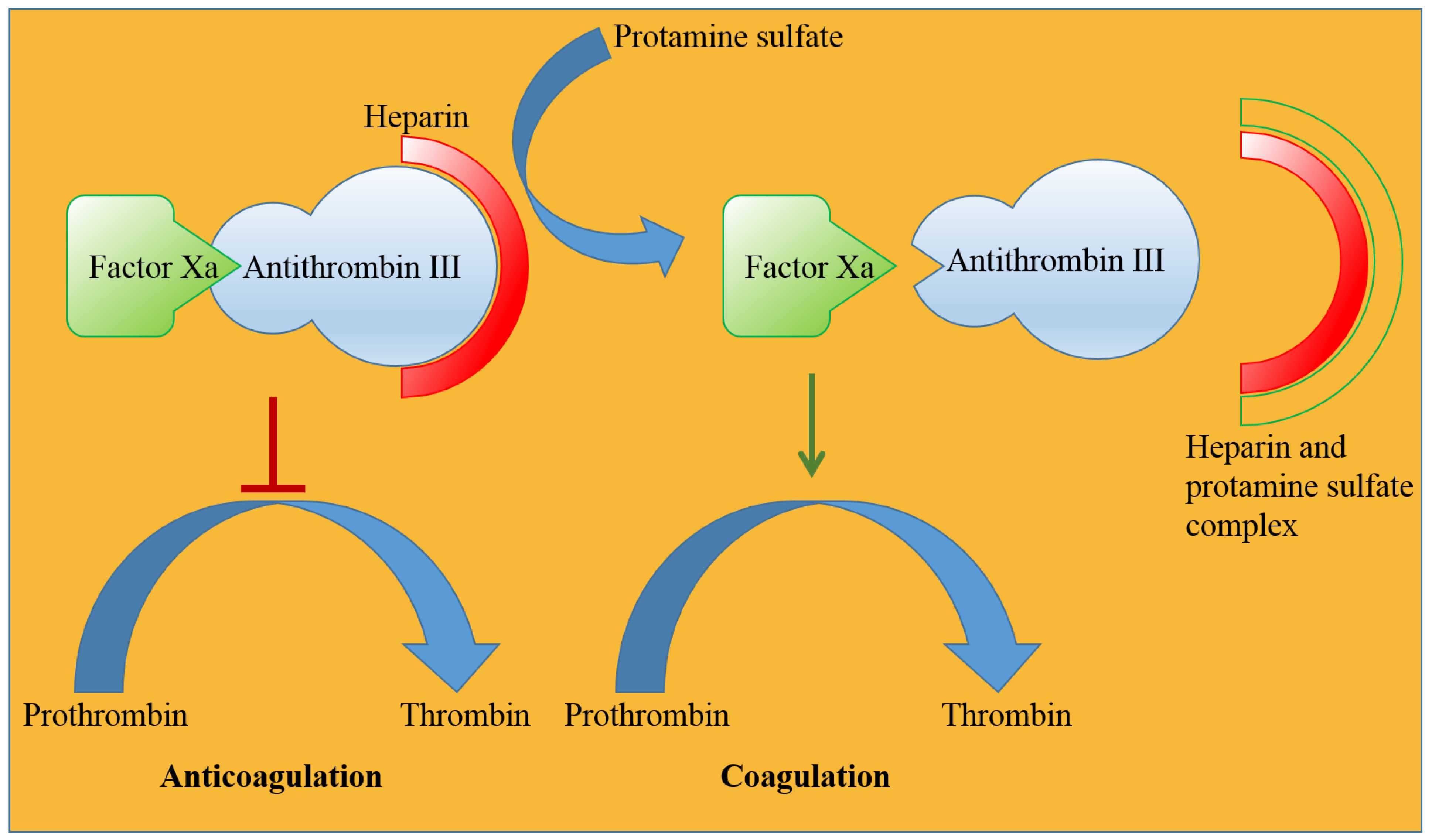
Figure 9. Cationic protamine sulfate reverses the anticoagulant effect of heparin by forming an ionic complex with heparin, which facilitates strong binding of factor Xa and weak binding of antithrombin and restores the natural coagulation process.
5.4. Keyhole Limpet Hemocyanin (KLH)
KLH is a large barrel-shaped, multi-subunit, oxygen-carrying metalloprotein present in the hemolymph of the marine mollusk Megathura crenulata (Figure 10A) [165]. KLH in itself is not a drug, but it is approved (as mentioned in DrugBank) for the preparation of therapeutic agents for the treatment of cancer and other immunological diseases by eliciting an appropriate immune response in human patients [166]. KLH is an immunomodulator and has made a valuable contribution to immunological research and the global immunopharmaceutical market for more than 50 years since its astounding immunostimulatory properties were documented in experimental animals and even in humans [167][168][169][170].
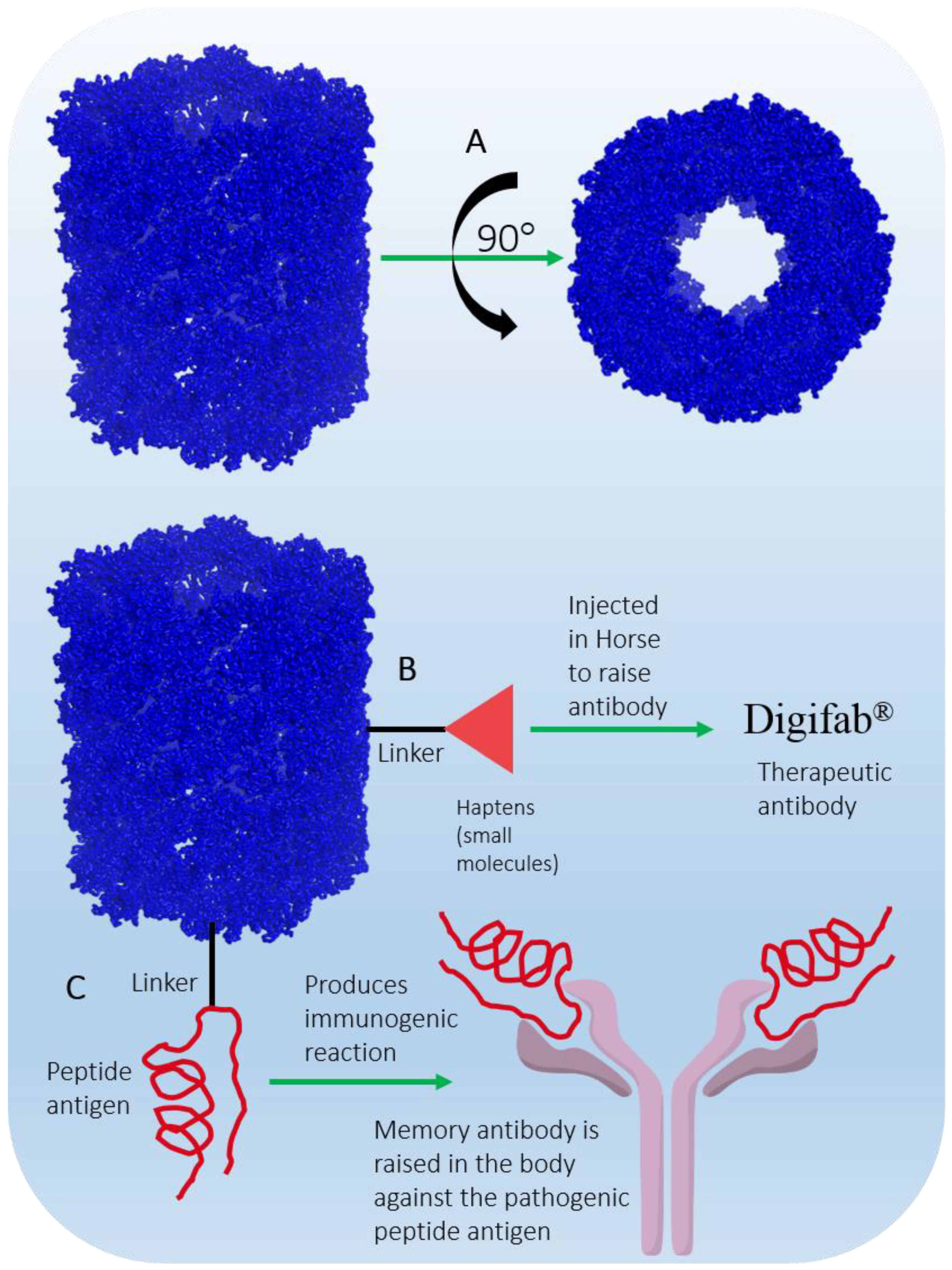
Figure 10. (A) KLH barrel-shaped representation drawn using cryo-electron microscopy 3D coordinates (PDB ID 4BED). (B) KLH as a hapten conjugate. (C) KLH as a vaccine carrier.
KLH is present in two isoforms, KLH1 and KLH2, with monomeric molecular masses of 390 kDa and 360 kDa, respectively [171]. KLH is composed of 20 monomers, where each monomer contains 7–8 functional domains. Each domain contains two copper ions (Cu2+) and binds one O2 molecule. The gigantic didecameric structure is heavily glycosylated, which makes it suitable for desirable hapten conjugation (Figure 10B), and it is used as a vaccine carrier molecule (Figure 10C). Due to its complex glycosylation and large size, laboratory synthesis is nearly impossible; therefore, commercial production is achieved by purification from the hemolymph of the animal [172]. KLH is immunogenic but does not elicit adverse immune responses in humans; hence, it is safe to use as an immunomodulator [173]. Immucothel® from Biosyn Corporation is a commercial subunit product that has been used as a building block for the reassociation of KLH in the presence of appropriate ions. Immucothel® was approved in 1997 for the treatment of bladder cancer and is marketed in the Netherlands, Austria, Argentina, and South Korea [173][174]. Vacmune® is another immunocyanin commercial product from Biosyn Corporation used as a protein carrier for vaccine development (Figure 10B) [175]. Commercially available KLH is often used to generate antibodies against small molecular moieties present in large complexes. The antibodies generated have been used as therapeutic agents in the production of the approved and marketed antibody drug Digifab® (ovine Digoxin Immune Fab), BTG International Inc., (London, UK) 2017 (Figure 10B) [176]. Other commercial products generated from KLH that have been used to analyze antibody titers in various immunological assays are ImmuneActivator™ (PerImmune, Rockville, MD, USA), KLH-ImmuneActivator® (Organon Technica, Durham, USA), and ImmuneActivator™ (Intracel, Rockville, MD, USA). A more comprehensive list of commercial KHL products used for immunotoxicological studies has been discussed in detail by Swaminathan et al. [177]. Active research to explore and optimize the immunostimulatory and immunomodulatory properties of KLH, which are of a very complex nature, may achieve therapy for diseases such as cancer in the future.
6. Fish Oil and Its Components
Fishes are well known for their health benefits worldwide. Fish accumulate essential fatty acids such as omega-3 fatty acids (primarily synthesized in marine algae) in their body through the marine food chain. Epidemiological studies have shown that people who consume fish regularly are at lower risk of coronary heart disease mortality than those who consume fish less often or consume none [178][179][180][181][182]. The health benefits associated with omega-3 fatty acids are mainly attributed to (a) alpha linolenic acid (ALA, C18:3, n-3), mostly of plant origin, (b) eicosapentaenoic acid (EPA, C20:5, n-3), which are mostly of marine origin, and (c) docosahexaenoic acid (DHA, C22:6, n-3), which are mostly of marine origin (Figure 11).
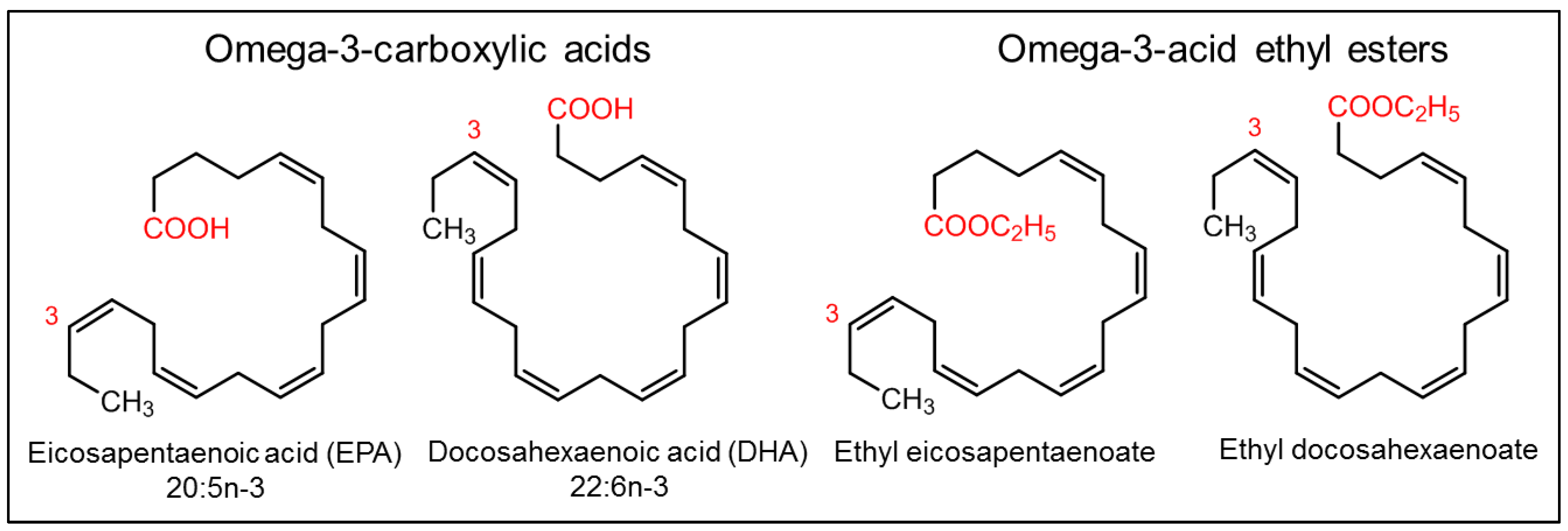
Figure 11. Omega-3 fatty acids and their derivatives.
Many of the fish oil components and their derivatives are approved by the US FDA as nutraceuticals and are commercially available on the market today (Figure 11) [183], including (a) omega-3-acid ethyl esters, marketed as Lovaza® (2004, GlaxoSmithKline, Brentford, UK) and in European countries as Omacor® (2004, GlaxoSmithKline, Brentford, UK) and Omtryg® (2004, Trygg Pharma, Oslo, Norway), (b) icosapent ethyl, marketed as Vascepa® (2012, Amarin Pharma, Inc., Dublin, Ireland), (c) omega-3-carboxylic acids, marketed as Epanova® (2014, AstraZeneca Pharmaceuticals LP, London, UK), and (d) fish oil triglycerides, marketed as Omegaven® (2018, Fresenius Kabi, Fresenius Kabi, Bad Homburg, Germany) [184][185].
Omega-3-acid ethyl esters, icosapent ethyl, and omega-3-carboxylic acids are all approved for the treatment of hypertriglyceridemia (triglycerides > 150 mg/dL) and severe hypertriglyceridemia (triglycerides > 500 mg/dL) [186][187][188]. These substances differ in the form of fatty acids and their amount in each 1 g capsule. Lovaza®, Omacor®, and Omtryg® contain a mixture of ethyl esters of EPA and DHA. Vascepa® contains the ethyl ester of EPA as a major ingredient [188]. Epanova® contains a mixture of carboxylic acids of EPA and DHA. The bioavailability of forms of EPA and DHA was highest in Vacepa®. Other inactive ingredients are also present in these formulations, such as tocopherol, gelatin, glycerin, maltitol, sorbitol, and purified water. These formulations help to reduce triglyceride and very-low-density lipoprotein levels (Figure 12). The mechanism underlying these effects is not very well understood, but the potential mechanisms proposed to date include the inhibition of diacylglycerol acyltransferase, increased activity of plasma lipoprotein lipase, a decrease in lipogenesis, and an increase in β-oxidation in the liver (Figure 12) [184][189].
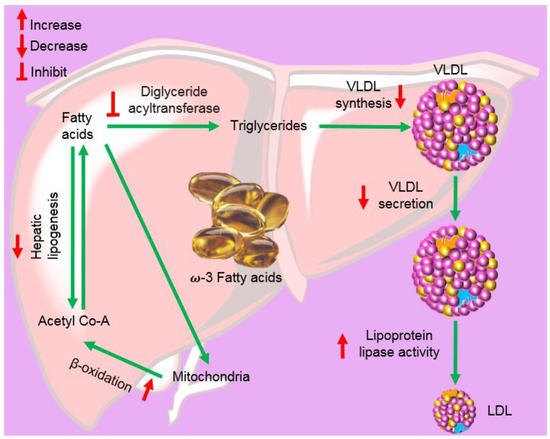
Figure 12. Mechanism via which omega-3 fatty acids lower triglycerides and the conversion of very-low-density lipoprotein (VLDL) into low-density lipoprotein (LDL).
Omegaven® is a fish oil-based emulsion approved for the treatment of parenteral nutrition-associated cholestasis (PNAC) in pediatric patients as a source of calories and fatty acids. Major risk factors for the development of PNAC are premature birth and low birth weight [190]. The drug is administered intravenously as an emulsion injection. The drug is not known to prevent PNAC. The mechanism underlying the success of the treatment is unknown, as researchers have not determined whether the anti-inflammatory effect or the effect of the ratio of omega-3/omega-6 fatty acid components in the drug is responsible. Experts recommend routine laboratory tests to monitor the levels of essential fatty acids when treating patients, as they often drop below normal levels [191].
References
- Kremer, W.B. Drugs five years later: Cytarabine. Ann. Intern. Med. 1975, 82, 684–688.
- Drugs. Cytarabine. Available online: https://www.drugs.com/monograph/cytarabine.html (accessed on 30 June 2022).
- Heuser, M.; Ofran, Y.; Boissel, N.; Mauri, S.B.; Craddock, C.; Janssen, J.; Wierzbowska, A.; Buske, C. Acute myeloid leukaemia in adult patients: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 697–712.
- Chu, M.Y.; Fischer, G. A proposed mechanism of action of 1-β-d-arabinofuranosyl-cytosine as an inhibitor of the growth of leukemic cells. Biochem. Pharmacol. 1962, 11, 423–430.
- Furth, J.; Cohen, S.S. Inhibition of mammalian DNA polymerase by the 5’-triphosphate of 1-β-D-arabinofuranosylcytosine and the 5’-triphosphate of 9-β-D-arabinofuranosyladenine. Cancer Res. 1968, 28, 2061–2067.
- Graham, F.; Whitmore, G. The effect of 1-β-D-arabinofuranosylcytosine on growth, viability, and DNA synthesis of mouse L-cells. Cancer Res. 1970, 30, 2627–2635.
- Plunkett, W.; Gandhi, V. Evolution of the arabinosides and the pharmacology of fludarabine. Drugs 1994, 47, 30–38.
- Chu, M.-Y. Incorporation of arabinosyl cytosine into 2–7S ribonucleic acid and cell death. Biochem. Pharmacol. 1971, 20, 2057–2063.
- Creasey, W.A.; Papac, R.J.; Markiw, M.E.; Calabresi, P.; Welch, A.D. Biochemical and pharmacological studies with 1-β-d-arabinofuranosylcytosine in man. Biochem. Pharmacol. 1966, 15, 1417–1428.
- Benedict, W.F.; Harris, N.; Karon, M. Kinetics of 1-β-D-arabinofuranosylcytosine-induced chromosome breaks. Cancer Res. 1970, 30, 2477–2483.
- Kihlman, B.; Nichols, W.W.; Levan, A. The effect of deoxyadenosine and cytosine arabinoside on the chromosomes of human leukocytes in vitro. Hereditas 1963, 50, 139–143.
- Camiener, G.W.; Smith, C.G. Studies of the enzymatic deamination of cytosine arabinoside—I: Enzyme distribution and species specificity. Biochem. Pharmacol. 1965, 14, 1405–1416.
- Mulligan, L.; Mellett, L. Comparative Metabolism of Cytosine Arabinoside and Inhibition of Deamination by Tetrahydrouridine. Pharmacologist 1968, 10, 167.
- Glantz, M.J.; LaFollette, S.; Jaeckle, K.A.; Shapiro, W.; Swinnen, L.; Rozental, J.R.; Phuphanich, S.; Rogers, L.R.; Gutheil, J.C.; Batchelor, T. Randomized trial of a slow-release versus a standard formulation of cytarabine for the intrathecal treatment of lymphomatous meningitis. J. Clin. Oncol. 1999, 17, 3110–3116.
- Zimm, S.; Collins, J.M.; Miser, J.; Chatterji, D.; Poplack, D.G. Cytosine arabinoside cerebrospinal fluid kinetics. Clin. Pharmacol. Ther. 1984, 35, 826–830.
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 2684–2692.
- Lim, W.-S.; Tardi, P.G.; Dos Santos, N.; Xie, X.; Fan, M.; Liboiron, B.D.; Huang, X.; Harasym, T.O.; Bermudes, D.; Mayer, L.D. Leukemia-selective uptake and cytotoxicity of CPX-351, a synergistic fixed-ratio cytarabine: Daunorubicin formulation, in bone marrow xenografts. Leuk. Res. 2010, 34, 1214–1223.
- Rein, L.A.; Rizzieri, D.A. Clinical potential of elacytarabine in patients with acute myeloid leukemia. Ther. Adv. Hematol. 2014, 5, 211–220.
- Baker, W.J.; Royer, G.L., Jr.; Weiss, R.B. Cytarabine and neurologic toxicity. J. Clin. Oncol. 1991, 9, 679–693.
- Tilly, H.; Castaigne, S.; Bordessoule, D.; Casassus, P.; Le Prise, P.; Tertian, G.; Desablens, B.; Henry-Amar, M.; Degos, L. Low-dose cytarabine versus intensive chemotherapy in the treatment of acute nonlymphocytic leukemia in the elderly. J. Clin. Oncol. 1990, 8, 272–279.
- Privat de Garilhe, M. Effect de deux nucleosides de l’arabinose sur la multiplication des virus de l’herpes et de la vaccine en culture cellulaire. CR Acad. Sci. 1964, 259, 2725–2728.
- Sidwell, R.W.; Arnett, G.; Dixon, G.J. Effect of selected biologically active compounds on in vitro cytomegalovirus (CMV) infections. In Proceedings of the 7th Interscience Conference on Antimicrobial Agents and Chemotherapy, Chicago, IL, USA, 28–29 October 1967.
- Miller, F.; Dixon, G.; Ehrlich, J.; Sloan, B.J.; McLean, I. Antiviral activity of 9 beta D arabinofuranosyladenîne. I. Cell culture studies. Antimicrob. Agents Chemother. 1969, 8, 136–147.
- Lee, W.W.; Benitez, A.; Goodman, L.; Baker, B. Potential anticancer agents. 1 xl. synthesis of the β-anomer of 9-(d-arabinofuranosyl)-adenine. J. Am. Chem. Soc. 1960, 82, 2648–2649.
- Buchanan, R.A.; Hess, F. Vidarabine (Vira-A®): Pharmacology and clinical experience. Pharmacol. Ther. 1980, 8, 143–171.
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265.
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747.
- Plunkett, W.; Chubb, S.; Alexander, L.; Montgomery, J.A. Comparison of the toxicity and metabolism of 9-β-D-arabinofuranosyl-2-fluoroadenine and 9-β-D-arabinofuranosyladenine in human lymphoblastoid cells. Cancer Res. 1980, 40, 2349–2355.
- Plunkett, W.; Benjamin, R.S.; Keating, M.J.; Freireich, E.J. Modulation of 9-β-D-arabinofuranosyladenine 5’-triphosphate and deoxyadenosine triphosphate in leukemic cells by 2′-deoxycoformycin during therapy with 9-β-D-arabinofuranosyladenine. Cancer Res. 1982, 42, 2092–2096.
- Plunkett, W.; Feun, L.G.; Benjamin, R.S.; Keating, M.; Freireich, E.J. “Modulation of Vidarabine Metabolism by 2’-Deoxycoformycin for Therapy of Acute Leukemia” Conference on 2′-Deoxycoformycin--Current Status and Future Directions, US Department of Health and Human Services, Public Health Service; National Institutes of Health: Washington, DC, USA, 1984; Volume 2, pp. 23–27.
- Montgomery, J.A.; Hewson, K. Synthesis of potential anticancer agents. X. 2-fluoroadenosine1. J. Am. Chem. Soc. 1957, 79, 4559.
- Rodriguez, G. Fludarabine phosphate. Investig. New Drugs 1994, 12, 75–92.
- Keating, M.; Kantarjian, H.; Talpaz, M.; Redman, J.; Koller, C.; Barlogie, B.; Velasquez, W.; Plunkett, W.; Freireich, E.J.; McCredie, K. Fludarabine: A new agent with major activity against chronic lymphocytic leukemia. Blood 1989, 74, 19–25.
- Rai, K.R.; Peterson, B.L.; Appelbaum, F.R.; Kolitz, J.; Elias, L.; Shepherd, L.; Hines, J.; Threatte, G.A.; Larson, R.A.; Cheson, B.D. Fludarabine compared with chlorambucil as primary therapy for chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1750–1757.
- McLaughlin, P.; Hagemeister, F.B.; Romaguera, J.E.; Sarris, A.H.; Pate, O.; Younes, A.; Swan, F.; Keating, M.; Cabanillas, F. Fludarabine, mitoxantrone, and dexamethasone: An effective new regimen for indolent lymphoma. J. Clin. Oncol. 1996, 14, 1262–1268.
- Thompson, P.A.; Tam, C.S.; O’Brien, S.M.; Wierda, W.G.; Stingo, F.; Plunkett, W.; Smith, S.C.; Kantarjian, H.M.; Freireich, E.J.; Keating, M.J. Fludarabine, cyclophosphamide, and rituximab treatment achieves long-term disease-free survival in IGHV-mutated chronic lymphocytic leukemia. Blood 2016, 127, 303–309.
- Carpenter, J., Jr.; Vogel, C.; Wang, G.; Raney, M. Phase II evaluation of fludarabine in patients with metastatic breast cancer: A southeastern cancer study group trial. Cancer Treat. Rep. 1986, 70, 1235–1236.
- Taylor, S.; Eyre, H. Randomized phase II trials of acivicin (AT-125, NSC 163501) and fludarabine (2-fluoro-ara-AMP, NSC 312887) (2FLAMP) in recurrent malignant gliomas: A SWOG study. Proc. Annu. Meet Am. Soc. Clin. Oncol. 1987, 6, 71.
- Taylor, S.A.; Crowley, J.; Vogel, F.S.; Townsend, J.J.; Eyre, H.J.; Jaeckle, K.A.; Hynes, H.E.; Guy, J.T. Phase II evaluation of fludarabine phosphate in patients with central nervous system tumors. Investig. New Drugs 1991, 9, 195–197.
- Weiss, G.; Metch, B.; Von Hoff, D.; Taylor, S.; Saiers, J. Phase II trial of fludarabine phosphate in patients with head and neck cancer: A southwest oncology group study. Cancer Treat. Rep. 1987, 71, 1313–1314.
- Casper, E.S.; Mittelman, A.; Kelson, D.; Young, C.W. Phase I clinical trial of fludarabine phosphate (F-ara-AMP). Cancer Chemother. Pharmacol. 1985, 15, 233–235.
- Hutton, J.J.; Von Hoff, D.D.; Kuhn, J.; Phillips, J.; Hersh, M.; Clark, G. Phase I clinical investigation of 9-β-d-arabinofuranosyl-2-fluoroadenine 5’-monophosphate (NSC 312887), a new purine antimetabolite. Cancer Res. 1984, 44, 4183–4186.
- Cheson, B.D.; Bennett, J.M.; Rai, K.R.; Grever, M.R.; Kay, N.E.; Schiffer, C.A.; Oken, M.M.; Keating, M.J.; Boldt, D.H.; Kempin, S.J.; et al. Guidelines for clinical protocols for chronic lymphocytic leukemia: Recommendations of the National Cancer Institute-sponsored working group. Am. J. Hematol. 1988, 29, 152–163.
- Hiddemann, W.; Rottmann, R.; Wörmann, B.; Thiel, A.; Essink, M.; Ottensmeier, C.; Freund, M.; Büchner, T.; van de Loo, J. Treatment of advanced chronic lymphocytic leukemia by fludarabine. Ann. Hematol. 1991, 63, 1–4.
- Hurst, P.G.; Habib, M.P.; Garewal, H.; Bluestein, M.; Paquin, M.; Greenberg, B.R. Pulmonary toxicity associated with fludarabine monophosphate. Investig. New Drugs 1987, 5, 207–210.
- Chun, H.; Leyland-Jones, B.; Caryk, S.; Hoth, D. Central nervous system toxicity of fludarabine phosphate. Cancer Treat. Rep. 1986, 70, 1225–1228.
- Warrell, R.P., Jr.; Berman, E. Phase I and II study of fludarabine phosphate in leukemia: Therapeutic efficacy with delayed central nervous system toxicity. J. Clin. Oncol. 1986, 4, 74–79.
- Jensen, K.; L’Aurelle, A.J.; Jacobson, P.A.; Kachler, S.; Kirstein, M.N.; Lamba, J.; Klotz, K.-N. Cytotoxic purine nucleoside analogues bind to A1, A2A, and A3 adenosine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 519–525.
- Ribeiro, J.; Sebastiao, A.; de Mendonça, A. Adenosine receptors in the nervous system: Pathophysiological implications. Prog. Neurobiol. 2002, 68, 377–392.
- Helman, D.L., Jr.; Byrd, J.C.; Ales, N.C.; Shorr, A.F. Fludarabine-related pulmonary toxicity: A distinct clinical entity in chronic lymphoproliferative syndromes. Chest 2002, 122, 785–790.
- Frederiksen, S. Specificity of adenosine deaminase toward adenosine and 2’-deoxyadenosine analogues. Arch. Biochem. Biophys. 1966, 113, 383–388.
- Avramis, V.I.; Champagne, J.; Sato, J.; Krailo, M.; Ettinger, L.J.; Poplack, D.G.; Finklestein, J.; Reaman, G.; Hammond, G.D.; Holcenberg, J.S. Pharmacology of fludarabine phosphate after a phase I/II trial by a loading bolus and continuous infusion in pediatric patients. Cancer Res. 1990, 50, 7226–7231.
- Malspeis, L.; Grever, M.R.; Staubus, A.; Young, D. Pharmacokinetics of 2-F-ara-A (9-beta-D-arabinofuranosyl-2-fluoroadenine) in cancer patients during the phase I clinical investigation of fludarabine phosphate. Semin. Oncol. 1990, 17, 18–32.
- Plunkett, W.; Huang, P.; Gandhi, V. Metabolism and action of fludarabine phosphate. Semin. Oncol. 1990, 17, 3–17.
- Plunkett, W.; Saunders, P.P. Metabolism and action of purine nucleoside analogs. Pharmacol. Ther. 1991, 49, 239–268.
- Danhauser, L.; Plunkett, W.; Keating, M.; Cabanillas, F. 9-β-D-Arabinofuranosyl-2-fluoroadenine 5’-monophosphate pharmacokinetics in plasma and tumor cells of patients with relapsed leukemia and lymphoma. Cancer Chemother. Pharmacol. 1986, 18, 145–152.
- Huang, P.; Chubb, S.; Plunkett, W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. a mechanism for cytotoxicity. J. Biol. Chem. 1990, 265, 16617–16625.
- Spriggs, D.; Robbins, G.; Mitchell, T.; Kufe, D. Incorporation of 9-β-D-arabinofuranosyl-2-fluoroadenine into HL-60 cellular RNA and DNA. Biochem. Pharmacol. 1986, 35, 247–252.
- Krenitsky, T.A.; Koszalka, G.W.; Tuttle, J.V.; Rideout, J.L.; Elion, G.B. An enzymic synthesis of purine D-arabinonucleosides. Carbohydr. Res. 1981, 97, 139–146.
- Lambe, C.U.; Averett, D.R.; Paff, M.T.; Reardon, J.E.; Wilson, J.G.; Krenitsky, T.A. 2-Amino-6-methoxypurine arabinoside: An agent for T-cell malignancies. Cancer Res. 1995, 55, 3352–3356.
- Gandhi, V.; Keating, M.J.; Bate, G.; Kirkpatrick, P. Nelarabine. Nat. Rev. Drug Discov. 2006, 5, 17–18.
- Mitchell, B.S.; Mejias, E.; Daddona, P.E.; Kelley, W.N. Purinogenic immunodeficiency diseases: Selective toxicity of deoxyribonucleosides for T cells. Proc. Natl. Acad. Sci. USA 1978, 75, 5011–5014.
- Rodriguez, C.O., Jr.; Stellrecht, C.M.; Gandhi, V. Mechanisms for T-cell selective cytotoxicity of arabinosylguanine. Blood 2003, 102, 1842–1848.
- DeAngelo, D.J.; Yu, D.; Johnson, J.L.; Coutre, S.E.; Stone, R.M.; Stopeck, A.T.; Gockerman, J.P.; Mitchell, B.S.; Appelbaum, F.R.; Larson, R.A. Nelarabine induces complete remissions in adults with relapsed or refractory T-lineage acute lymphoblastic leukemia or lymphoblastic lymphoma: Cancer and leukemia group B study 19801. Blood 2007, 109, 5136–5142.
- Gandhi, V.; Tam, C.; O’Brien, S.; Jewell, R.C.; Rodriguez, C.O., Jr.; Lerner, S.; Plunkett, W.; Keating, M.J. Phase I trial of nelarabine in indolent leukemias. J. Clin. Oncol. 2008, 26, 1098–1105.
- Mishchenko, N.P.; Fedoreev, S.A.; Bagirova, V.L. Histochrome: A new original domestic drug. Pharm. Chem. J. 2003, 37, 48–52.
- Artyukov, A.A.; Zelepuga, E.A.; Bogdanovich, L.N.; Lupach, N.M.; Novikov, V.L.; Rutckova, T.A.; Kozlovskaya, E.P. Marine polyhydroxynaphthoquinone, echinochrome a: Prevention of atherosclerotic inflammation and probable molecular targets. J. Clin. Med. 2020, 9, 1494.
- Park, J.H.; Lee, N.K.; Lim, H.J.; Mazumder, S.; Kumar Rethineswaran, V.; Kim, Y.J.; Jang, W.B.; Ji, S.T.; Kang, S.; Kim, D.Y.; et al. Therapeutic cell protective role of histochrome under oxidative stress in human cardiac progenitor cells. Mar. Drugs 2019, 17, 368.
- Hirata, Y.; Uemura, D. Halichondrins-antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710.
- Bauer, A. Story of eribulin mesylate: Development of the longest drug synthesis. In Synthesis of Heterocycles in Contemporary Medicinal Chemistry; Springer: Berlin/Heidelberg, Germany, 2016; pp. 209–270.
- Seshadri, P.; Deb, B.; Kumar, P. Multifarious targets beyond microtubules-role of eribulin in cancer therapy. Front. Biosci. 2021, 13, 157–172.
- USFDA. Highlights of Prescribing Information: HALAVEN™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/201532lbl.pdf (accessed on 30 June 2022).
- Eisai (Global) Eisai Announces Canadian Approval of Its Anticancer Agent Halaven™. Available online: https://www.eisai.com/news/news201179.html (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: HALAVEN™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/201532s015lbl.pdf (accessed on 30 June 2022).
- Polastro, L.; Aftimos, P.G.; Awada, A. Eribulin mesylate in the management of metastatic breast cancer and other solid cancers: A drug review. Expert Rev. Anticancer. Ther. 2014, 14, 649–665.
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsky, B.M.; Palme, M.H.; et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of halichondrin B. Cancer Res. 2001, 61, 1013–1021.
- Ganjoo, K.N.; Patel, S.R. Trabectedin: An anticancer drug from the sea. Expert Opin. Pharmacother. 2009, 10, 2735–2743.
- Guan, Y.; Sakai, R.; Rinehart, K.; Wang, A. Molecular and crystal structures of ecteinascidins: Potent antitumor compounds from the caribbean tunicate ecteinascidia turbinata. J. Biomol. Struct. Dyn. 1993, 10, 793–818.
- Corey, E.J.; Gin, D.Y.; Kania, R.S. Enantioselective total synthesis of ecteinascidin 743. J. Am. Chem. Soc. 1996, 118, 9202–9203.
- He, W.; Zhang, Z.; Ma, D. A scalable total synthesis of the antitumor agents ET-743 and lurbinectedin. Angew. Chem. Int. Ed. 2019, 58, 3972–3975.
- EMA. Public Summary of Opinion on Orphan Designation: Trabectedin for the Treatment of Ovarian Cancer. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/03/171-public-summary-positive-opinion-orphan-designation-trabectedin-treatment-ovarian-cancer_en.pdf (accessed on 30 June 2022).
- EMA. Yondelis. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/yondelis (accessed on 30 June 2022).
- De Sanctis, R.; Marrari, A.; Santoro, A. Trabectedin for the treatment of soft tissue sarcomas. Expert Opin. Pharmacother. 2016, 17, 1569–1577.
- EMA. Committee for Medicinal Products for Human Use Post-Authorisation Summary of Positive Opinion for Yondelis. Available online: https://www.ema.europa.eu/en/documents/smop/chmp-post-authorisation-summary-positive-opinion-yondelis-24-september-2009_en.pdf (accessed on 30 June 2022).
- Zijoo, R.; von Mehren, M. Efficacy of trabectedin for the treatment of liposarcoma. Expert Opin. Pharmacother. 2016, 17, 1953–1962.
- USFDA. Highlights of Prescribing Information: YONDELIS™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/207953s000lbl.pdf (accessed on 30 June 2022).
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for soft tissue sarcoma: Current status and future perspectives. Adv. Ther. 2016, 33, 1055–1071.
- Grosso, F.; D’Ambrosio, L.; Zucchetti, M.; Ibrahim, T.; Tamberi, S.; Matteo, C.; Rulli, E.; Comandini, D.; Palmerini, E.; Baldi, G.G.; et al. Pharmacokinetics, safety, and activity of trabectedin as first-line treatment in elderly patients who are affected by advanced sarcoma and are unfit to receive standard chemotherapy: A phase 2 study (TR1US study) from the Italian sarcoma group. Cancer 2020, 126, 4726–4734.
- Beumer, J.H.; Rademaker-Lakhai, J.M.; Rosing, H.; Hillebrand, M.J.; Bosch, T.M.; Lopez-Lazaro, L.; Schellens, J.H.; Beijnen, J.H. Metabolism of trabectedin (ET-743, Yondelis) in patients with advanced cancer. Cancer Chemother. Pharmacol. 2007, 59, 825–837.
- Gago, F.; Hurley, L.H. Devising a structural basis for the potent cytotoxic effects of Ecteinascidin 743. In Small Molecule DNA and RNA Binders: From Synthesis to Nulceic Acid; The Royal Society of Chemistry: London, UK, 2002; pp. 643–675.
- Jimeno, J.; Maki, R.G.; Casali, P.; Faircloth, G.; Martinez, N.; Nieto, A.; Cañigueral, S.; Rinehart, K. Therapeutic impact of ET-743 (yondelis; trabectidin), a new marine-derived compound, in sarcoma. Curr. Opin. Orthop. 2003, 14, 419–428.
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K.W. DNA sequence-and structure-selective alkylation of guanine N2 in the DNA minor groove by ecteinascidin 743, a potent antitumor compound from the Caribbean tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309.
- Erba, E.; Bergamaschi, D.; Bassano, L.; Damia, G.; Ronzoni, S.; Faircloth, G.; d’Incalci, M. Ecteinascidin-743 (ET-743), a natural marine compound, with a unique mechanism of action. Eur. J. Cancer 2001, 37, 97–105.
- Takebayashi, Y.; Pourquier, P.; Zimonjic, D.B.; Nakayama, K.; Emmert, S.; Ueda, T.; Urasaki, Y.; Kanzaki, A.; Akiyama, S.-I.; Popescu, N.; et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat. Med. 2001, 7, 961–966.
- Soares, D.G.; Escargueil, A.E.; Poindessous, V.; Sarasin, A.; de Gramont, A.; Bonatto, D.; Henriques, J.A.P.; Larsen, A.K. Replication and homologous recombination repair regulate DNA double-strand break formation by the antitumor alkylator ecteinascidin 743. Proc. Natl. Acad. Sci. USA 2007, 104, 13062–13067.
- Liguori, M.; Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages as incessant builders and destroyers of the cancer stroma. Cancers 2011, 3, 3740–3761.
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013, 23, 249–262.
- D’incalci, M.; Badri, N.; Galmarini, C.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650.
- Leal, J.; Martínez-Díez, M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.; Negri, A.; Gago, F.; Guillén-Navarro, M.; Avilés, P. PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Br. J. Pharmacol. 2010, 161, 1099–1110.
- Newswire, P.R. Jazz Pharmaceuticals Announces U.S. FDA Accelerated Approval of zepzelca™ (lurbinectedin) for the Treatment of Metastatic Small Cell Lung Cancer. Available online: https://www.prnewswire.com/news-releases/jazz-pharmaceuticals-announces-us-fda-accelerated-approval-of-zepzelca-lurbinectedin-for-the-treatment-of-metastatic-small-cell-lung-cancer-301077082.html (accessed on 30 June 2022).
- Baena, J.; Modrego, A.; Zeaiter, A.; Kahatt, C.; Alfaro, V.; Jimenez-Aguilar, E.; Mazarico, J.M.; Paz-Ares, L. Lurbinectedin in the treatment of relapsed small cell lung cancer. Future Oncol. 2021, 17, 2279–2289.
- USFDA. Highlights of Prescribing Information: ZEPZELCA™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213702s000lbl.pdf (accessed on 30 June 2022).
- Trigo, J.; Subbiah, V.; Besse, B.; Moreno, V.; López, R.; Sala, M.A.; Peters, S.; Ponce, S.; Fernández, C.; Alfaro, V.; et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: A single-arm, open-label, phase 2 basket trial. Lancet Oncol. 2020, 21, 645–654.
- Fernandez-Teruel, C.; Gonzalez, I.; Trocóniz, I.F.; Lubomirov, R.; Soto, A.; Fudio, S. Population-pharmacokinetic and covariate analysis of lurbinectedin (PM01183), a new RNA polymerase II Inhibitor, in pooled phase I/II trials in patients with cancer. Clin. Pharmacokinet. 2019, 58, 363–374.
- Elez, M.E.; Tabernero, J.; Geary, D.; Macarulla, T.; Kang, S.P.; Kahatt, C.; Pita, A.S.-M.; Teruel, C.F.; Siguero, M.; Cullell-Young, M. First-in-human phase I study of Lurbinectedin (PM01183) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2205–2214.
- Moneo, V.; Martínez, P.; de Castro, B.; Cascajares, S.; Avila, S.; Garcia-Fernandez, L.F.; Galmarini, C.M. Abstract A174: Comparison of the antitumor activity of trabectedin, lurbinectedin, zalypsis and PM00128 in a panel of human cells deficient in transcription/NER repair factors. Am. Assoc. Cancer Res. 2017, 5, 628–638.
- Nuñez, G.S.; Robles CM, G.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.-M. Lurbinectedin specifically triggers the degradation of phosphorylated RNA polymerase II and the formation of DNA breaks in cancer cells. Mol. Cancer Ther. 2016, 15, 2399–2412.
- Pernice, T.; Bishop, A.G.; Guillen, M.J.; Cuevas, C.; Aviles, P. Development of a liquid chromatography/tandem mass spectrometry assay for the quantification of PM01183 (lurbinectedin), a novel antineoplastic agent, in mouse, rat, dog, Cynomolgus monkey and mini-pig plasma. J. Pharm. Biomed. Anal. 2016, 123, 37–41.
- Romano, M.; Frapolli, R.; Zangarini, M.; Bello, E.; Porcu, L.; Galmarini, C.M.; García-Fernández, L.F.; Cuevas, C.; Allavena, P.; Erba, E. Comparison of in vitro and in vivo biological effects of trabectedin, lurbinectedin (PM01183) and zalypsis®(PM00104). Int. J. Cancer 2013, 133, 2024–2033.
- Belgiovine, C.; Bello, E.; Liguori, M.; Craparotta, I.; Mannarino, L.; Paracchini, L.; Beltrame, L.; Marchini, S.; Galmarini, C.M.; Mantovani, A.; et al. Lurbinectedin reduces tumour-associated macrophages and the inflammatory tumour microenvironment in preclinical models. Br. J. Cancer 2017, 117, 628–638.
- Taniguchi, H.; Sen, T.; Rudin, C.M. Targeted therapies and biomarkers in small cell lung cancer. Front. Oncol. 2020, 10, 741.
- USFDA. Highlights of Prescribing Information: ADCETRIS™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125388_S056S078lbl.pdf (accessed on 30 June 2022).
- Horie, R.; Watanabe, T. Seminars in Immunology. In CD30: Expression and Function in Health and Disease; Elsevier: Amsterdam, The Netherlands, 1998; pp. 457–470.
- Van der Weyden, C.; Pileri, S.; Feldman, A.; Whisstock, J.; Prince, H. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603.
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862.
- Hansen, H.P.; Trad, A.; Dams, M.; Zigrino, P.; Moss, M.; Tator, M.; Schön, G.; Grenzi, P.C.; Bachurski, D.; Aquino, B. CD30 on extracellular vesicles from malignant Hodgkin cells supports damaging of CD30 ligand-expressing bystander cells with brentuximab-vedotin, in vitro. Oncotarget 2016, 7, 30523.
- Amaya, M.; Jimeno, A.; Kamdar, M. Polatuzumab vedotin to treat relapsed or refractory diffuse large B-cell lymphoma, in combination with bendamustine plus rituximab. Drugs Today 2020, 56, 287–294.
- Malecek, M.-K.; Watkins, M.P.; Bartlett, N.L. Polatuzumab vedotin for the treatment of adults with relapsed or refractory diffuse large B-cell lymphoma. Expert Opin. Biol. Ther. 2021, 21, 831–839.
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586.
- International Non-Hodgkin’s Lymphoma Prognostic Factors Project, A predictive model for aggressive non-Hodgkin’s lymphoma. N. Engl. J. Med. 1993, 329, 987–994.
- USFDA. Clinical Review: Polatuzumab Vedotin™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/761121Orig1s000MedR.pdf (accessed on 30 June 2022).
- USFDA. Highlights of Prescribing Information: POLIVY™. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 30 June 2022).
- FDA. Padcev®. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-enfortumab-vedotin-ejfv-locally-advanced-or-metastatic-urothelial-cancer (accessed on 30 June 2022).
- FDA Approved Drug Products: Padcev (Enfortumab Vedotin-Ejfv) for IV Injection. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf (accessed on 30 June 2022).
- Hanna, K.S. Clinical overview of enfortumab vedotin in the management of locally advanced or metastatic urothelial carcinoma. Drugs 2020, 80, 1–7.
- McGregor, B.A.; Sonpavde, G. Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial carcinoma. Expert Opin. Investig. Drugs 2019, 28, 821–826.
- FDA. Blenrep®. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-granted-accelerated-approval-belantamab-mafodotin-blmf-multiple-myeloma (accessed on 30 June 2022).
- FDA Approved Drug Products: Blenrep Belantaman Mafodotin-Blmf Intravenous Injection. Available online: http://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761158s006lbl.pdf (accessed on 30 June 2022).
- McMillan, A.; Warcel, D.; Popat, R. Antibody-drug conjugates for multiple myeloma. Expert Opin. Biol. Ther. 2021, 21, 889–901.
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody-drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37.
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238.
- Rinehart, K.L., Jr.; Gloer, J.B.; Cook, J.C., Jr.; Mizsak, S.A.; Scahill, T.A. Structures of the didemnins, antiviral and cytotoxic depsipeptides from a caribbean tunicate. J. Am. Chem. Soc. 1981, 103, 1857–1859.
- Rinehart, K.L.; Kishore, V.; Bible, K.C.; Sakai, R.; Sullins, D.W.; Li, K.-M. Didemnins and tunichlorin: Novel natural products from the marine tunicate trididemnum solidum. J. Nat. Pproducts 1988, 51, 1–21.
- TGA, A. Australian Public Assessment Report for Plitidepsin. Available online: https://www.tga.gov.au/sites/default/files/auspar-plitidepsin-190513.pdf (accessed on 30 June 2022).
- BioWorld. EU Court Sides with Pharmamar in Aplidin Approval Dispute. Available online: https://www.bioworld.com/articles/499498-eu-court-sides-with-pharmamar-in-aplidin-approval-dispute (accessed on 30 June 2022).
- White, K.M.; Rosales, R.; Yildiz, S.; Kehrer, T.; Miorin, L.; Moreno, E.; Jangra, S.; Uccellini, M.B.; Rathnasinghe, R.; Coughlan, L.; et al. Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 2021, 371, 926–931.
- Gomez, J.; Extremera, S.; Nieto, A. Overall survival (OS) results of randomized phase III study (ADMYRE trial) of plitidepsin and dexamethasone (DXM) vs. DXM alone in patients with relapsed/refractory multiple myeloma (RRMM): Evaluation of the crossover impact. Am. Soc. Clin. Oncol. J. 2018, 36, 8018.
- Spicka, I.; Ocio, E.M.; Oakervee, H.E.; Greil, R.; Banh, R.H.; Huang, S.-Y.; D’Rozario, J.M.; Dimopoulos, M.A.; Martínez, S.; Extremera, S.; et al. Randomized phase III study (ADMYRE) of plitidepsin in combination with dexamethasone vs. dexamethasone alone in patients with relapsed/refractory multiple myeloma. Ann. Hematol. 2019, 98, 2139–2150.
- Ribrag, V.; Caballero, D.; Fermé, C.; Zucca, E.; Arranz, R.; Briones, J.; Gisselbrecht, C.; Salles, G.; Gianni, A.M.; Gomez, H. Multicenter phase II study of plitidepsin in patients with relapsed/refractory non-Hodgkin’s lymphoma. Haematologica 2013, 98, 357–363.
- Muñoz-Alonso, M.J.; González-Santiago, L.; Martínez, T.; Losada, A.; Galmarini, C.M.; Muñoz, A. The mechanism of action of plitidepsin. Curr. Opin. Investig. Drugs 2009, 10, 536–542.
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martín, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253–264.
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68.
- Jin, A.-H.; Muttenthaler, M.; Dutertre, S.; Himaya, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Conotoxins: Chemistry and biology. Chem. Rev. 2019, 119, 11510–11549.
- Kaas, Q.; Westermann, J.-C.; Craik, D.J. Conopeptide characterization and classifications: An analysis using conoserver. Toxicon 2010, 55, 1491–1509.
- Kumar, P.S.; Kumar, D.S.; Umamaheswari, S. A perspective on toxicology of conus venom peptides. Asian Pac. J. Trop. Med. 2015, 8, 337–351.
- Bäckryd, E. Do the potential benefits outweigh the risks? An update on the use of ziconotide in clinical practice. Eur. J. Pain 2018, 22, 1193–1202.
- Bozorgi, H.; Motaghi, E.; Zamani, M.; Ghavimi, R. Neuronal calcium channels blocker, ziconotide (ɷ-conotoxin MVIIA), reverses morphine withdrawal-induced memory impairments via alteration in hippocampal NMDA receptor expression in rats. Toxin Rev. 2018, 39, 1–10.
- Wermeling, D.; Drass, M.; Ellis, D.; Mayo, M.; McGuire, D.; O’Connell, D.; Hale, V.; Chao, S. Pharmacokinetics and pharmacodynamics of intrathecal ziconotide in chronic pain patients. J. Clin. Pharmacol. 2003, 43, 624–636.
- Schmidtko, A.; Lötsch, J.; Freynhagen, R.; Geisslinger, G. Ziconotide for treatment of severe chronic pain. Lancet 2010, 375, 1569–1577.
- Smith, H.S.; Deer, T.R. Safety and efficacy of intrathecal ziconotide in the management of severe chronic pain. Ther. Clin. Risk Manag. 2009, 5, 521–534.
- Nair, A.; Poornachand, A.; Kodisharapu, P. Ziconotide: Indications, adverse effects, and limitations in managing refractory chronic pain. Indian J. Palliat. Care 2018, 24, 118–119.
- Deer, T.R.; Prager, J.; Levy, R.; Rathmell, J.; Buchser, E.; Burton, A.; Caraway, D.; Cousins, M.; de Andrés, J.; Diwan, S.; et al. Polyanalgesic consensus conference 2012: Recommendations for the management of pain by intrathecal (intraspinal) drug delivery: Report of an interdisciplinary expert panel. Neuromodulation 2012, 15, 436–466.
- Deer, T.; Hagedorn, J.M. How has ziconotide impacted non-cancer pain management? Expert Opin. Pharmacother. 2020, 21, 507–511.
- Matis, G.; de Negri, P.; Dupoiron, D.; Likar, R.; Zuidema, X.; Rasche, D. Intrathecal pain management with ziconotide: Time for consensus? Brain Behav. 2021, 11, e02055.
- Jorpes, E.; Thaning, T. Neutralisation of action of heparin by protamine. Lancet 1939, 234, 975–976.
- Sokolowska, E.; Kalaska, B.; Miklosz, J.; Mogielnicki, A. The toxicology of heparin reversal with protamine: Past, present and future. Expert Opin. Drug Metab. Toxicol. 2016, 12, 897–909.
- Drugs. Protamine Sulfate. Available online: https://www.drugs.com/international/protamine-sulfate.html (accessed on 30 June 2022).
- Lindblad, B. Protamine sulphate: A review of its effects: Hypersensitivity and toxicity. Eur. J. Vasc. Surg. 1989, 3, 195–201.
- Sorgi, F.; Bhattacharya, S.; Huang, L. Protamine sulfate enhances lipid-mediated gene transfer. Gene Ther. 1997, 4, 961–968.
- DeLucia, A., 3rd; Wakefield, T.W.; Kadell, A.; Wrobleski, S.K.; VanDort, M.; Stanley, J.C. Tissue distribution, circulating half-life, and excretion of intravenously administered protamine sulfate. J. Am. Soc. Artif. Intern. Organs 1993, 39, M715–M718.
- Blajchman, M. An overview of the mechanism of action of antithrombin and its inherited deficiency states. Blood Coagul. Fibrinolysis 1994, 5, S5–S11.
- Hirsh, J.; Warkentin, T.E.; Dalen, J.E.; Deykin, D.; Poller, L. Heparin: Mechanism of action, pharmacokinetics, dosing considerations, monitoring, efficacy, and safety. Chest 1995, 108, 258S–275S.
- Okajima, Y.; Kanayama, S.; Maeda, Y.; Urano, S.; Kitani, T.; Watada, M.; Nakagawa, M.; Ijichi, H. Studies on the neutralizing mechanism of antithrombin activity of heparin by protamine. Thromb. Res. 1981, 24, 21–29.
- Medpagetoday. FDA Clears Andexanet Alfa for Rapid NOAC Reversal. Available online: https://www.medpagetoday.com/cardiology/prevention/72701 (accessed on 30 June 2022).
- Klhsite. About KLH. Available online: https://www.klhsite.com/about-klh/ (accessed on 30 June 2022).
- DrugBank. Keyhole Limpet Hemocyanin. Available online: https://go.drugbank.com/drugs/DB05299 (accessed on 30 June 2022).
- Weigle, W.O. Immunochemical properties of hemocyanin. Immunochemistry 1964, 1, 295–302.
- Curtis, J.; Hersh, E.M.; Harris, J.; McBride, C.; Freireich, E.J. The human primary immune response to keyhole limpet haemocyanin: Interrelationships of delayed hypersensitivity, antibody response and in vitro blast transformation. Clin. Exp. Immunol. 1970, 6, 473–491.
- Curtis, J.E.; Hersh, E.M.; Butler, W.T.; Rossen, R.D. Antigen dose in the human immune response: Dose-response relationships in the human immune response to keyhole limpet hemocyanin. J. Lab. Clin. Med. 1971, 78, 61–69.
- Herscowitz, H.; Harold, W.; Stavitsky, A. Immunochemical and immunogenic properties of a purified keyhole limpet haemocyanin. Immunology 1972, 22, 51–61.
- Söhngen, S.M.; Stahlmann, A.; Harris, J.R.; Müller, S.A.; Engel, A.; Markl, J. Mass determination, subunit organization and control of oligomerization states of keyhole limpet hemocyanin (KLH). Eur. J. Biochem. 1997, 248, 602–614.
- Harris, J.; Markl, J. Keyhole limpet hemocyanin (KLH): A biomedical review. Micron 1999, 30, 597–623.
- Nseyo, U.O.; Lamm, D.L. Seminars in Surgical Oncology. In Immunotherapy of Bladder Cancer; Wiley Online Library: Hoboken, NJ, USA, 1997; pp. 342–349.
- Biosyn. IMMUCOTHEL™. Available online: https://biosyncorp.com/klh/ (accessed on 30 June 2022).
- Biosyn. VACMUNE™. Available online: https://biosyncorp.com/vacmuner/ (accessed on 30 June 2022).
- International, B. DigiFab. Available online: https://digifab.health/en-us (accessed on 30 June 2022).
- Swaminathan, A.; Lucas, R.M.; Dear, K.; McMichael, A.J. Keyhole limpet haemocyanin–a model antigen for human immunotoxicological studies. Br. J. Clin. Pharmacol. 2014, 78, 1135–1142.
- Shekelle, R.B.; Missell, L.; Paul, O.; Shryock, A.M.; Stamler, J.; Vollset, S.E.; Heuch, I.; Bjelke, E.; Curb, J.D.; Reed, D.M. Fish consumption and mortality from coronary heart disease. N. Engl. J. Med. 1985, 313, 820–824.
- Dolecek, T.; Granditis, G. Dietary polyunsaturated fatty acids and mortality in the multiple risk factor intervention trial (MRFIT). World Rev. Nutr. Diet. 1991, 66, 205–216.
- Kromhout, D.; Feskens, E.J.; Bowles, C.H. The protective effect of a small amount of fish on coronary heart disease mortality in an elderly population. Int. J. Epidemiol. 1995, 24, 340–345.
- Zhang, J.; Sasaki, S.; Amano, K.; Kesteloot, H. Fish consumption and mortality from all causes, ischemic heart disease, and stroke: An ecological study. Prev. Med. 1999, 28, 520–529.
- Kris-Etherton, P.M.; Harris, W.S.; Appel, L.J. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation 2002, 106, 2747–2757.
- Kumar, N.G.; Contaifer, D.; Madurantakam, P.; Carbone, S.; Price, E.T.; van Tassell, B.; Brophy, D.F.; Wijesinghe, D.S. Dietary bioactive fatty acids as modulators of immune function: Implications on human health. Nutrients 2019, 11, 2974.
- Maki, K.C.; Johns, C.; Harris, W.S.; Puder, M.; Freedman, S.D.; Thorsteinsson, T.; Daak, A.; Rabinowicz, A.L.; Sancilio, F.D. Bioequivalence demonstration for Ω-3 acid ethyl Ester formulations: Rationale for modification of current guidance. Clin. Ther. 2017, 39, 652–658.
- Guthrie, G.; Burrin, D. Impact of parenteral lipid emulsion components on cholestatic liver disease in neonates. Nutrients 2021, 13, 508.
- Saremi, A.; Arora, R. The utility of omega-3 fatty acids in cardiovascular disease. Am. J. Ther. 2009, 16, 421–436.
- Ballantyne, C.M.; Braeckman, R.A.; Soni, P.N. Icosapent ethyl for the treatment of hypertriglyceridemia. Expert Opin. Pharmacother. 2013, 14, 1409–1416.
- Bazarbashi, N.; Miller, M. Icosapent ethyl: Drug profile and evidence of reduced residual cardiovascular risk in patients with statin-managed LDL-C cholesterol. Expert Rev. Cardiovasc. Ther. 2020, 18, 175–180.
- Backes, J.; Anzalone, D.; Hilleman, D.; Catini, J. The clinical relevance of omega-3 fatty acids in the management of hypertriglyceridemia. Lipids Health Dis. 2016, 15, 1–12.
- Touloukian, R.J.; Seashore, J.H. Hepatic secretory obstruction with total parenteral nutrition in the infant. J. Pediatric Surg. 1975, 10, 353–360.
- Rangel, S.J.; Calkins, C.M.; Cowles, R.A.; Barnhart, D.C.; Huang, E.Y.; Abdullah, F.; Arca, M.J.; Teitelbaum, D.H. Parenteral nutrition–associated cholestasis: An American pediatric surgical association outcomes and clinical trials committee systematic review. J. Pediatric Surg. 2012, 47, 225–240.
More
Information
Subjects:
Marine & Freshwater Biology; Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.9K
Revisions:
3 times
(View History)
Update Date:
29 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No