 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mark William Nachtigal | -- | 2035 | 2022-08-19 13:18:50 | | | |
| 2 | Vivi Li | Meta information modification | 2035 | 2022-08-22 04:03:55 | | | | |
| 3 | Vivi Li | -12 word(s) | 2023 | 2022-08-23 10:48:38 | | |
Video Upload Options
Recurrent epithelial ovarian cancer (EOC) coincident with chemotherapy resistance remains the main contributor to patient mortality. There is an ongoing investigation to enhance patient progression-free and overall survival with novel chemotherapeutic delivery, such as the utilization of antiangiogenic medications, PARP inhibitors, or immune modulators. Glycosylated antitumor ether lipids (GAELs) are synthetic glycerolipids capable of killing established human epithelial cell lines from a wide variety of human cancers, including EOC cell lines representative of different EOC histotypes. Importantly, GAELs kill high-grade serous ovarian cancer (HGSOC) cells isolated from the ascites of chemotherapy-sensitive and chemotherapy-resistant patients grown as monolayers of spheroid cultures. In addition, GAELs were well tolerated by experimental animals (mice) and were capable of reducing tumor burden and blocking ascites formation in an OVCAR-3 xenograft model. Overall, GAELs show great promise as adjuvant therapy for EOC patients with or without chemotherapy resistance.
1. Introduction
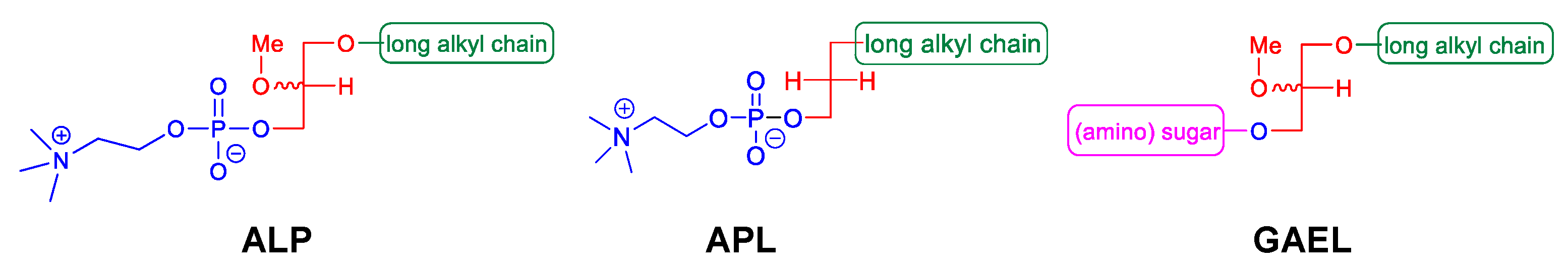
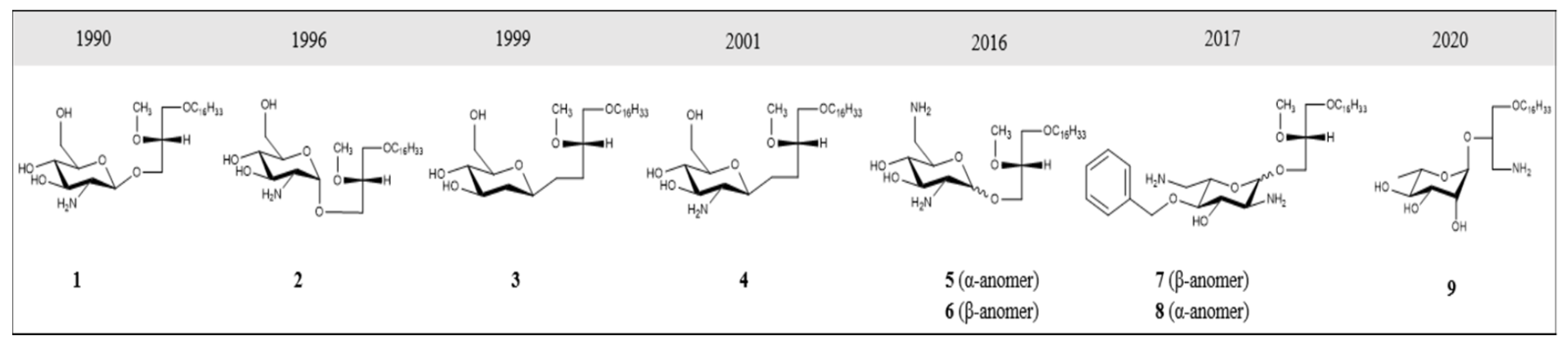
2. GAELs: How Did We Get Here?
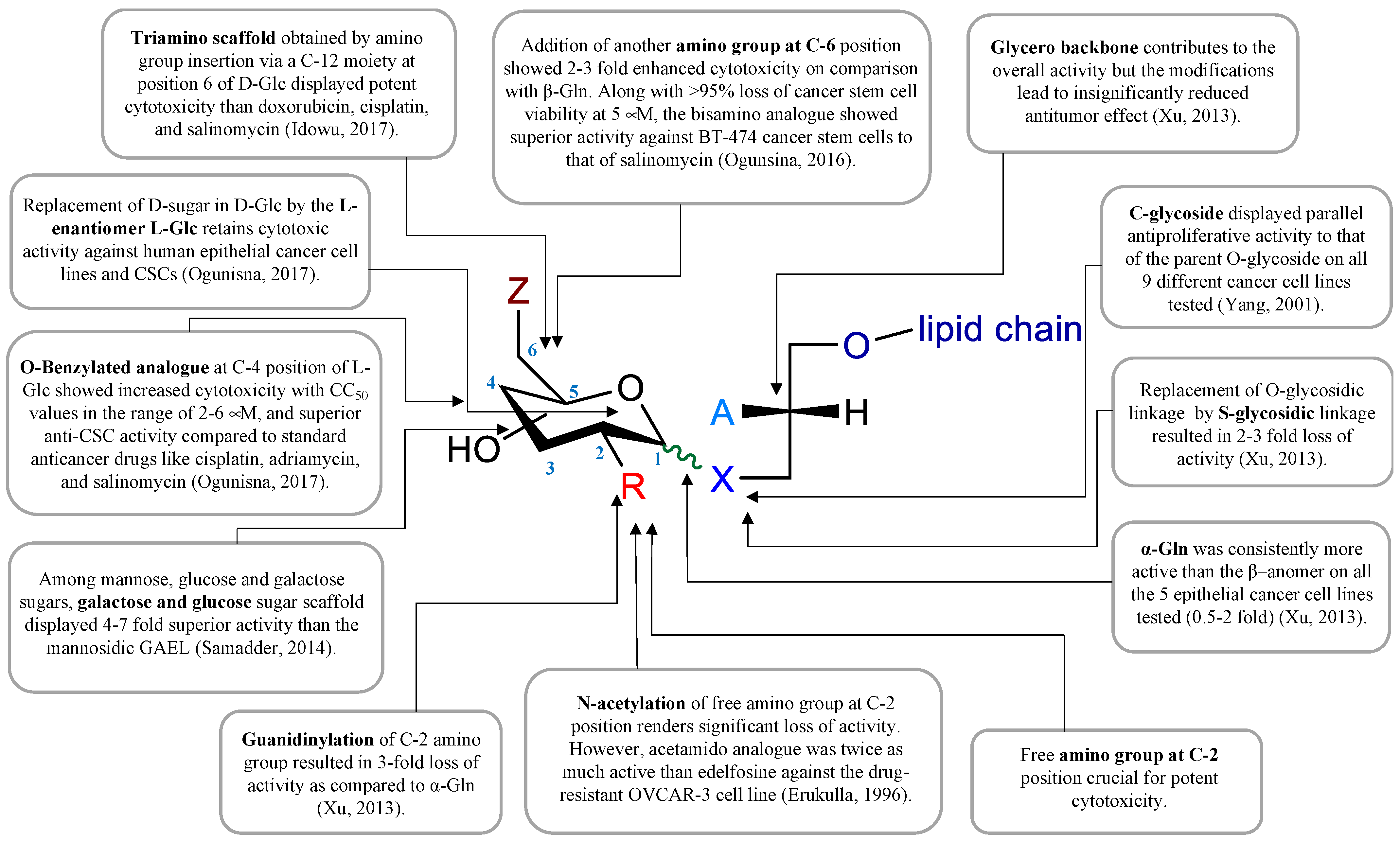
3. GAEL Structure–Activity Relationship
References
- CCS. Canadian Cancer Statistics 2021. 2021. Available online: https://cdn.cancer.ca/-/media/files/research/cancer-statistics/2021-statistics/2021-pdf-en-final.pdf?rev=2b9d2be7a2d34c1dab6a01c6b0a6a32d&hash=01DE85401DBF0217F8B64F2B7DF43986 (accessed on 14 June 2022).
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568.
- Buys, S.S.; Partridge, E.; Black, A.; Johnson, C.C.; Lamerato, L.; Isaacs, C. Effect of screening on ovarian cancer mortality: The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Randomized Controlled Trial. JAMA 2011, 305, 2295–2303.
- Gilbert, L.; Basso, O.; Sampalis, J.; Karp, I.; Martins, C.; Feng, J. Assessment of symptomatic women for early diagnosis of ovarian cancer: Results from the prospective DOvE pilot project. Lancet Oncol. 2012, 13, 285–291.
- Goff, B.A.; Mandel, L.S.; Drescher, C.W.; Urban, N.; Gough, S.; Schurman, K.M. Development of an ovarian cancer symptom index: Possibilities for earlier detection. Cancer 2007, 109, 221–227.
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2016, 387, 945–956.
- Lu, K.H.; Skates, S.; Hernandez, M.A.; Bedi, D.; Bevers, T.; Leeds, L. A 2-stage ovarian cancer screening strategy using the Risk of Ovarian Cancer Algorithm (ROCA) identifies early-stage incident cancers and demonstrates high positive predictive value. Cancer 2013, 119, 3454–3461.
- Altman, A.D.; Lambert, P.; Love, A.J.; Turner, D.; Lotocki, R.; Dean, E. Examining the Effects of Time to Diagnosis, Income, Symptoms, and Incidental Detection on Overall Survival in Epithelial Ovarian Cancer: Manitoba Ovarian Cancer Outcomes (MOCO) Study Group. Int. J. Gynecol. Cancer 2017, 27, 1637–1644.
- Nagle, C.M.; Francis, J.E.; Nelson, A.E.; Zorbas, H.; Luxford, K.; de Fazio, A. Reducing time to diagnosis does not improve outcomes for women with symptomatic ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2011, 29, 2253–2258.
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N. Engl. J. Med. 1996, 334, 1–6.
- Piccart, M.J.; Bertelsen, K.; James, K.; Cassidy, J.; Mangioni, C.; Simonsen, E. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: Three-year results. J. Natl. Cancer Inst. 2000, 92, 699–708.
- International Collaborative Ovarian Neoplasm Group. Paclitaxel plus carboplatin versus standard chemotherapy with either single-agent carboplatin or cyclophosphamide, doxorubicin, and cisplatin in women with ovarian cancer: The ICON3 randomised trial. Lancet 2002, 360, 505–515.
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: A Gynecologic Oncology Group study. J. Clin. Oncol. 2003, 21, 3194–3200.
- Sherman-Baust, C.A.; Becker, K.G.; Wood, W.H., III; Zhang, Y.; Morin, P.J. Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J. Ovarian Res. 2011, 4, 21.
- Suh, D.H.; Kim, M.K.; No, J.H.; Chung, H.H.; Song, Y.S. Metabolic approaches to overcoming chemoresistance in ovarian cancer. Ann. N. Y. Acad. Sci. 2011, 1229, 53–60.
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257.
- May, T.; Altman, A.; McGee, J.; Lu, L.; Xu, W.; Lane, K. Examining Survival Outcomes of 852 Women with Advanced Ovarian Cancer: A Multi-institutional Cohort Study. Int. J. Gynecol. Cancer 2018, 28, 925–931.
- Vergote, I.; Trope, C.G.; Amant, F.; Kristensen, G.B.; Ehlen, T.; Johnson, N. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med. 2010, 363, 943–953.
- Katsumata, N.; Yasuda, M.; Takahashi, F.; Isonishi, S.; Jobo, T.; Aoki, D. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: A phase 3, open-label, randomised controlled trial. Lancet 2009, 374, 1331–1338.
- Pignata, S.; Scambia, G.; Katsaros, D.; Gallo, C.; Pujade-Lauraine, E.; De Placido, S. Carboplatin plus paclitaxel once a week versus every 3 weeks in patients with advanced ovarian cancer (MITO-7): A randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 396–405.
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43.
- Van Driel, W.J.; Koole, S.N.; Sikorska, K.; Schagen van Leeuwen, J.H.; Schreuder, H.W.R.; Hermans, R.H.M. Hyperthermic Intraperitoneal Chemotherapy in Ovarian Cancer. N. Engl. J. Med. 2018, 378, 230–240.
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483.
- Chan, J.K.; Brady, M.F.; Penson, R.T.; Huang, H.; Birrer, M.J.; Walker, J.L. Weekly vs. Every-3-Week Paclitaxel and Carboplatin for Ovarian Cancer. N. Engl. J. Med. 2016, 374, 738–748.
- Oza, A.M.; Cook, A.D.; Pfisterer, J.; Embleton, A.; Ledermann, J.A.; Pujade-Lauraine, E. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): Overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015, 16, 928–936.
- Walker, J.L.; Brady, M.F.; Wenzel, L.; Fleming, G.F.; Huang, H.Q.; DiSilvestro, P.A. Randomized Trial of Intravenous Versus Intraperitoneal Chemotherapy Plus Bevacizumab in Advanced Ovarian Carcinoma: An NRG Oncology/Gynecologic Oncology Group Study. J. Clin. Oncol. 2019, 37, 1380–1390.
- DiSilvestro, P.; Colombo, N.; Harter, P.; Gonzalez-Martin, A.; Ray-Coquard, I.; Coleman, R.L. Maintenance Treatment of Newly Diagnosed Advanced Ovarian Cancer: Time for a Paradigm Shift? Cancers 2021, 13, 5756.
- Markman, M.; Liu, P.Y.; Wilczynski, S.; Monk, B.; Copeland, L.J.; Alvarez, R.D. Phase III randomized trial of 12 versus 3 months of maintenance paclitaxel in patients with advanced ovarian cancer after complete response to platinum and paclitaxel-based chemotherapy: A Southwest Oncology Group and Gynecologic Oncology Group trial. J. Clin. Oncol. 2003, 21, 2460–2465.
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1721–1731.
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402.
- Konstantinopoulos, P.A.; Lheureux, S.; Moore, K.N. PARP Inhibitors for Ovarian Cancer: Current Indications, Future Combinations, and Novel Assets in Development to Target DNA Damage Repair. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e116–e131.
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392.
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861.
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428.
- Leary, A.; Tan, D.; Ledermann, J. Immune checkpoint inhibitors in ovarian cancer: Where do we stand? Ther. Adv. Med. Oncol. 2021, 13, 17588359211039899.
- Arnold, M.; Rutherford, M.J.; Bardot, A.; Ferlay, J.; Andersson, T.M.; Myklebust, T.A. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995-2014 (ICBP SURVMARK-2): A population-based study. Lancet Oncol. 2019, 20, 1493–1505.
- Cabasag, C.J.; Butler, J.; Arnold, M.; Rutherford, M.; Bardot, A.; Ferlay, J. Exploring variations in ovarian cancer survival by age and stage (ICBP SurvMark-2): A population-based study. Gynecol. Oncol. 2020, 157, 234–244.
- Coleman, M.P.; Forman, D.; Bryant, H.; Butler, J.; Rachet, B.; Maringe, C. Cancer survival in Australia, Canada, Denmark, Norway, Sweden, and the UK, 1995–2007 (the International Cancer Benchmarking Partnership): An analysis of population-based cancer registry data. Lancet 2011, 377, 127–138.
- Lee, J.Y.; Kim, S.; Kim, Y.T.; Lim, M.C.; Lee, B.; Jung, K.W. Changes in ovarian cancer survival during the 20 years before the era of targeted therapy. BMC Cancer 2018, 18, 601.
- Lin, J.J.; Egorova, N.; Franco, R.; Prasad-Hayes, M.; Bickell, N.A. Ovarian Cancer Treatment and Survival Trends Among Women Older Than 65 Years of Age in the United States, 1995–2008. Obstet. Gynecol. 2016, 127, 81–89.
- Norell, C.H.; Butler, J.; Farrell, R.; Altman, A.; Bentley, J.; Cabasag, C.J. Exploring international differences in ovarian cancer treatment: A comparison of clinical practice guidelines and patterns of care. Int. J. Gynecol. Cancer 2020, 30, 1748–1756.
- Vogler, W.R.; Liu, J.; Volpert, O.; Ades, E.W.; Bouck, N. The anticancer drug edelfosine is a potent inhibitor of neovascularization in vivo. Cancer Investig. 1998, 16, 549–553.
- Croft, S.L.; Engel, J. Miltefosine—Discovery of the antileishmanial activity of phospholipid derivatives. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, S4–S8.
- Erdlenbruch, B.; Jendrossek, V.; Marx, M.; Hunold, A.; Eibl, H.; Lakomek, M. Antitumor effects of erucylphosphocholine on brain tumor cells in vitro and in vivo. Anticancer Res. 1998, 18, 2551–2557.
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a novel Akt inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110.
- Arthur, G.; Schweizer, F.; Ogunsina, M. Synthetic glycosylated ether glycerolipids as anticancer agents. In Carbohydrates in Drug Design and Discovery; Jiménez-Barbero, J., Cañada, F.J., Martin-Santamaria, S., Eds.; Royal Society of Chemistry: Cambridge, UK, 2015.
- Jahreiss, L.; Renna, M.; Bittman, R.; Arthur, G.; Rubinsztein, D.C. 1-O-Hexadecyl-2-O-methyl-3-O-(2′-acetamido-2′-deoxy-beta-D-glucopyranosyl)-sn-gly cerol (Gln) induces cell death with more autophagosomes which is autophagy-independent. Autophagy 2009, 5, 835–846.
- Moraya, A.I.; Ali, J.L.; Samadder, P.; Liang, L.; Morrison, L.C.; Werbowetski-Ogilvie, T.E. Novel glycolipid agents for killing cisplatin-resistant human epithelial ovarian cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 67.
- Nachtigal, M.W.; Musaphir, P.; Dhiman, S.; Altman, A.D.; Schweizer, F.; Arthur, G. Cytotoxic capacity of a novel glycosylated antitumor ether lipid in chemotherapy-resistant high grade serous ovarian cancer in vitro and in vivo. Transl. Oncol. 2021, 14, 101203.
- Ogunsina, M.; Samadder, P.; Idowu, T.; Arthur, G.; Schweizer, F. Design, synthesis and evaluation of cytotoxic properties of bisamino glucosylated antitumor ether lipids against cancer cells and cancer stem cells. Med. Chem. Comm. 2016, 7, 2100–2110.
- Samadder, P.; Bittman, R.; Byun, H.S.; Arthur, G. A glycosylated antitumor ether lipid kills cells via paraptosis-like cell death. Biochem. Cell Biol. 2009, 87, 401–414.
- Samadder, P.; Xu, Y.Z.; Schweizer, F.; Arthur, G. Cytotoxic properties of D-gluco-, D-galacto- and D-manno-configured 2-amino-2-deoxy-glycerolipids against epithelial cancer cell lines and BT-474 breast cancer stem cells. Eur. J. Med. Chem. 2014, 78, 225–235.
- Burdzy, K.; Munder, P.G.; Fischer, H.; Westphal, O. Increase in the Phagocytosis of Peritoneal Macrophages by Lysolecithin. Z. Naturforsch. B. 1964, 19, 1118–1120.
- Fischer, H. Lysolecithin and the Action of Complement. Ann. N. Y. Acad. Sci. 1964, 116, 1063–1070.
- Munder, P.G.; Modolell, M.; Ferber, E.; Fischer, H. Phospholipids in quartz-damaged macrophages. Biochem. Z. 1966, 344, 310–313.
- Arnold, D.; Weltzien, H.U.; Westphal, O. Concerning the synthesis of lysolecithin and its ether analogs. Justus Liebigs Ann. Chem. 1967, 709, 234–239.
- Eibl, H.; Westphal, O. Palmitoyl-propandiol-(1.3)-phosphorylcholine (2-desoxylysolecithin) and omega.omega’-alkanediol-analogs. Justus Liebigs Ann. Chem. 1967, 709, 244–245.
- Weltzien, H.U.; Westphal, O. O-methylated and O-acetylated lysolecithin. Justus Liebigs Ann. Chem. 1967, 709, 240–243.
- Munder, P.G.; Modolell, M.; Andreesen, R.; Weltzien, H.U.; Westphal, O. Lysophosphatidylcholine (Lysolecithin) and its Synthetic Analogues. Immunomodulating and Other Biologic Effects. In Immunostimulation; Chedid, L., Miescher, P.A., Mueller-Eberhard, H.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1980.
- Andreesen, R.; Modolell, M.; Weltzien, H.U.; Eibl, H.; Common, H.H.; Lohr, G.W. Selective destruction of human leukemic cells by alkyl-lysophospholipids. Cancer Res. 1978, 38, 3894–3899.
- Modolell, M.; Andreesen, R.; Pahlke, W.; Brugger, U.; Munder, P.G. Disturbance of phospholipid metabolism during the selective destruction of tumor cells induced by alkyl-lysophospholipids. Cancer Res. 1979, 39, 4681–4686.
- Tarnowski, G.S.; Mountain, I.M.; Stock, C.C.; Munder, P.G.; Weltzien, H.U.; Westphal, O. Effect of lysolecithin and analogs on mouse ascites tumors. Cancer Res. 1978, 38, 339–344.
- Berdel, W.E.; Bausert, W.R.; Weltzien, H.U.; Modolell, M.L.; Widmann, K.H.; Munder, P.G. The influence of alkyl-lysophospholipids and lysophospholipid-activated macrophages on the development of metastasis of 3-Lewis lung carcinoma. Eur. J. Cancer 1980, 16, 1199–1204.
- Berger, M.R.; Munder, P.G.; Schmahl, D.; Westphal, O. Influence of the alkyllysophospholipid ET-18-OCH3 on methylnitrosourea-induced rat mammary carcinomas. Oncology 1984, 41, 109–113.
- Helfman, D.M.; Barnes, K.C.; Kinkade, J.M.; Vogler, W.R.; Shoji, M.; Kuo, J.F. Phospholipid-Sensitive Ca-2+-Dependent Protein-Phosphorylation System in Various Types of Leukemic-Cells from Human Patients and in Human-Leukemic Cell Line-Hl60 and Line-K562, and Its Inhibition by Alkyl-Lysophospholipid. Cancer Res. 1983, 43, 2955–2961.
- Parker, J.; Daniel, L.W.; Waite, M. Evidence of Protein-Kinase-C Involvement in Phorbol Diester-Stimulated Arachidonic-Acid Release and Prostaglandin Synthesis. J. Biol. Chem. 1987, 262, 5385–5393.
- Daniel, L.W.; Etkin, L.A.; Morrison, B.T.; Parker, J.; Morris-Natschke, S.; Surles, J.R. Ether lipids inhibit the effects of phorbol diester tumor promoters. Lipids 1987, 22, 851–855.
- Vogler, W.R.; Whigham, E.; Bennett, W.D.; Olson, A.C. Effect of alkyl-lysophospholipids on phosphatidylcholine biosynthesis in leukemic cell lines. Exp. Hematol. 1985, 13, 629–633.
- Berdel, W.E.; Fromm, M.; Fink, U.; Pahlke, W.; Bicker, U.; Reichert, A. Cytotoxicity of thioether-lysophospholipids in leukemias and tumors of human origin. Cancer Res. 1983, 43, 5538–5543.
- Storme, G.A.; Berdel, W.E.; van Blitterswijk, W.J.; Bruyneel, E.A.; De Bruyne, G.K.; Mareel, M.M. Antiinvasive effect of racemic 1-O-octadecyl-2-O-methylglycero-3-phosphocholine on MO4 mouse fibrosarcoma cells in vitro. Cancer Res. 1985, 45, 351–357.
- Houlihan, W.J.; Lohmeyer, M.; Workman, P.; Cheon, S.H. Phospholipid antitumor agents. Med. Res. Rev. 1995, 15, 157–223.
- Smets, L.A.; Van Rooij, H.; Salomons, G.S. Signalling steps in apoptosis by ether lipids. Apoptosis 1999, 4, 419–427.
- Berdel, W.E.; Fink, U.; Rastetter, J. Clinical phase I pilot study of the alkyl lysophospholipid derivative ET-18-OCH3. Lipids 1987, 22, 967–969.
- Drings, P.; Günther, I.; Gatzemeier, U.; Ulbrich, F.; Khanavkar, B.; Schreml, W. Final Evaluation of a Phase II Study on the Effect of Edelfosine (an Ether Lipid) in Advanced Non-Small-Cell Bronchogenic Carcinoma. Onkologie 1992, 15, 375–382.
- Gajate, C.; Mollinedo, F. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH(3) (edelfosine), a proapoptotic agent in tumor cells. Curr. Drug Metab. 2002, 3, 491–525.
- Herrmann, D.B.; Neumann, H.A.; Berdel, W.E.; Heim, M.E.; Fromm, M.; Boerner, D. Phase I trial of the thioether phospholipid analogue BM 41.440 in cancer patients. Lipids 1987, 22, 962–966.
- Hilgard, P.; Stekar, J.; Voegeli, R.; Harleman, J.H. Experimental therapeutic studies with miltefosine in rats and mice. Prog. Exp. Tumor Res. 1992, 34, 116–130.
- Unger, C.; Eibl, H. Hexadecylphosphocholine: Preclinical and the first clinical results of a new antitumor drug. Lipids 1991, 26, 1412–1417.
- Jendrossek, V.; Erdlenbruch, B.; Hunold, A.; Kugler, W.; Eibl, H.; Lakomek, M. Erucylphosphocholine, a novel antineoplastic ether lipid, blocks growth and induces apoptosis in brain tumor cell lines in vitro. Int. J. Oncol. 1999, 14, 15–22.
- Kondapaka, S.B.; Singh, S.S.; Dasmahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003, 2, 1093–1103.
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633.
- Van Blitterswijk, W.J.; Verheij, M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim. Biophys. Acta 2013, 1831, 663–674.
- Keane, N.A.; Glavey, S.V.; Krawczyk, J.; O’Dwyer, M. AKT as a therapeutic target in multiple myeloma. Expert Opin. Ther. Targets 2014, 18, 897–915.
- Zentaris, A. Assessment of Efficacy and Safety of Perifosine, Bortezomib and Dexamethasone in Multiple Myeloma Patients. 2013. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01002248 (accessed on 14 June 2022).
- Weber, N.; Benning, H. Synthesis of Ether Glyceroglycolipids. Chem. Phys. Lipids 1986, 41, 93–100.
- Guivisdalsky, P.N.; Bittman, R.; Smith, Z.; Blank, M.L.; Snyder, F.; Howard, S. Synthesis and antineoplastic properties of ether-linked thioglycolipids. J. Med. Chem. 1990, 33, 2614–2621.
- Lu, X.L.; Rengan, K.; Bittman, R.; Arthur, G. The Alpha-Anomers and Beta-Anomers of 1-O-Hexadecyl-2-O-Methyl-3-S-Thioglucosyl-Sn-Glycerol Inhibit the Proliferation of Epithelial Cancer Cell-Lines. Oncol. Rep. 1994, 1, 933–936.
- Erukulla, R.K.; Zhou, X.; Samadder, P.; Arthur, G.; Bittman, R. Synthesis and evaluation of the antiproliferative effects of 1-O-hexadecyl-2-O-methyl-3-O-(2′-acetamido-2′-deoxy-beta-D-glucopyranosyl)-sn-glycerol and 1-O-hexadecyl-2-O-methyl-3-0-(2′-amino-2′-deoxy-beta-D-glucopyranosyl)-sn-glycerol on epithelial cancer cell growth. J. Med. Chem. 1996, 39, 1545–1548.
- Idowu, T.; Samadder, P.; Arthur, G.; Schweizer, F. Design, synthesis and antitumor properties of glycosylated antitumor ether lipid (GAEL)- chlorambucil-hybrids. Chem. Phys. Lipids 2016, 194, 139–148.
- Ogunsina, M.; Samadder, P.; Idowu, T.; Arthur, G.; Schweizer, F. Replacing D-Glucosamine with Its L-Enantiomer in Glycosylated Antitumor Ether Lipids (GAELs) Retains Cytotoxic Effects against Epithelial Cancer Cells and Cancer Stem Cells. J. Med. Chem. 2017, 60, 2142–2147.
- Xu, Y.Z.; Ogunsina, M.; Samadder, P.; Arthur, G.; Schweizer, F. StructureActivity Relationships of Glucosamine-Derived Glycerolipids: The Role of the Anomeric Linkage, the Cationic Charge and the Glycero Moiety on the Antitumor Activity. ChemMedChem 2013, 8, 511–520.
- Yang, G.L.; Franck, R.W.; Bittman, R.; Samadder, P.; Arthur, G. Synthesis and growth inhibitory properties of glucosamine-derived glycerolipids. Org. Lett. 2001, 3, 197–200.
- Yang, G.L.; Franck, R.W.; Byun, H.S.; Bittman, R.; Samadder, P.; Arthur, G. Convergent C-glycolipid synthesis via the Ramberg-Backlund reaction: Active antiproliferative glycolipids. Org. Lett. 1999, 1, 2149–2151.
- Idowu, T.; Samadder, P.; Arthur, G.; Schweizer, F. Amphiphilic Modulation of Glycosylated Antitumor Ether Lipids Results in a Potent Triamino Scaffold against Epithelial Cancer Cell Lines and BT474 Cancer Stem Cells. J. Med. Chem. 2017, 60, 9724–9738.
- Ogunsina, M.; Samadder, P.; Idowu, T.; Nachtigal, M.W.; Schweizer, F.; Arthur, G. Syntheses of L-Rhamnose linked amino glycerolipids and their cytotoxic activities against human cancer cells. Molecules 2020, 25, 566.

