Recurrent epithelial ovarian cancer (EOC) coincident with chemotherapy resistance remains the main contributor to patient mortality. There is an ongoing investigation to enhance patient progression-free and overall survival with novel chemotherapeutic delivery, such as the utilization of antiangiogenic medications, PARP inhibitors, or immune modulators. Our preclinical studies highlight a novel tool to combat chemotherapy-resistant human EOC. Glycosylated antitumor ether lipids (GAELs) are synthetic glycerolipids capable of killing established human epithelial cell lines from a wide variety of human cancers, including EOC cell lines representative of different EOC histotypes. Importantly, GAELs kill high-grade serous ovarian cancer (HGSOC) cells isolated from the ascites of chemotherapy-sensitive and chemotherapy-resistant patients grown as monolayers of spheroid cultures. In addition, GAELs were well tolerated by experimental animals (mice) and were capable of reducing tumor burden and blocking ascites formation in an OVCAR-3 xenograft model. Overall, GAELs show great promise as adjuvant therapy for EOC patients with or without chemotherapy resistance.
1. Introduction
Epithelial ovarian cancer (EOC) continues to be a lethal malignancy in women worldwide, and resistance to chemotherapy remains one of the main contributors to EOC patient morbidity and mortality
[1][2][1,2]. The early disease has a relatively good prognosis, and this fact led to screening and symptomatic-based studies
[3][4][5][6][7][3,4,5,6,7] in the hopes of detecting disease earlier and improving outcomes. Unfortunately, none of these studies have proven useful for increasing survival. Other studies examined the role of time-to-diagnosis and time-to-care and once again found no significant benefit for survival
[8][9][8,9]. Improved surgical techniques resulting in optimal tumor debulking, improved cytotoxic delivery methods, and novel non-cytotoxic chemotherapeutics are treatment modalities that have contributed to improved progression-free and overall survival in the EOC patient population in the past several decades. Herein,
reswe
archers review the development and utility of a novel glycolipid compound belonging to the glycosylated antitumor ether lipid (GAEL) class of drugs for the potential treatment of EOCs.
Historically, EOC was treated with multiple different agents until the late 1990s, when several studies confirmed the superiority of cisplatin plus paclitaxel as the basis of EOC chemotherapeutic treatments
[10][11][10,11]. Subsequent trials showed the equivalence of carboplatin to cisplatin, with decreased toxicity, leading to the fundamental base of modern cytotoxic therapy for EOC
[12][13][12,13]. While the majority of high-grade serous ovarian cancer (HGSOC) patients respond to initial chemotherapy (6–9 cycles of a platinum agent (carboplatin) and a taxane (paclitaxel)), up to 75% of EOC patients will relapse within 18 months with chemotherapy-resistant disease
[14][15][14,15]. Moreover, mucinous, clear cell and low-grade serous EOC histotypes are characterized by a poor response to chemotherapy. EOC therapy has continued to develop with changes in surgical approaches
[16][17][18][16,17,18], different approaches for dosing of chemotherapy (e.g., dose dense and dose intense)
[19][20][19,20], chemotherapy delivery (e.g., intraperitoneal and hyperthermic intraperitoneal)
[21][22][21,22], additional adjuvant agents (e.g., Bevacizumab)
[23][24][25][26][23,24,25,26], and maintenance therapy
[27][28][27,28]. The most promising recent advances in EOC management have surrounded advancements in poly-ADP ribose (PARP) inhibitor therapies, specifically for
BRCA1/
2 and homologous recombination-deficient (HRD) patients
[29][30][31][32][33][34][29,30,31,32,33,34]. Ongoing trials with immune modulators have not yet shown benefits for this patient population
[35]. Despite great advances in care, overall survival, although improved slightly, has not yet made drastic changes in several decades
[36][37][38][39][40][41][36,37,38,39,40,41]. Thus, additional treatments capable of preventing tumor growth and effectively killing chemoresistant EOC cells are desirable. The connection between translational research and clinical work will be critical in the advancement of gynecologic oncology and cancer therapy.
As part of
reseaour
chers' ongoing search for novel treatments for human epithelial cancers,
researcherswe developed glycosylated antitumor ether lipids (GAELs;
Figure 1) and demonstrated that GAELs exhibit anticancer activities in a diverse group of human cancer types in vitro. GAEL compounds are structurally related to antitumor ether lipids (AELs), including edelfosine
[42], miltefosine
[43], erucylphosphocholine
[44] and perifosine
[45] that have been tested as anticancer agents in clinical studies. GAELs would fall under the class of cytotoxic rather than targeted agents. GAELs are able to kill chemosensitive and chemoresistant human high-grade serous ovarian cancer (HGSOC) cell lines and patient samples grown as adherent or 3D spheroid cultures
[46][47][48][49][50][51][52][46,47,48,49,50,51,52]. Importantly, the GAEL mechanism of action is apoptosis-independent. This characteristic distinguishes GAELs from most of the currently used anticancer drugs that rely on apoptosis to kill tumor cells. Because cancer cells often overcome apoptosis induced by many cytotoxic agents, the addition of GAELs would be an excellent complementary treatment to overcome drug-resistance mechanisms. Moreover, as cellular genetic heterogeneity is observed in EOCs, the ability of GAELs to kill chemotherapy-sensitive and resistant cells would be an additional benefit of including GAELs as adjuvant treatment. This
re
ntryview introduces how GAELs were developed from observations of the biological activity of lysophosphatidylcholine (LPC), provides a brief overview of structure–activity relationship (SAR) studies, and highlights
researchers'our studies testing GAEL activity to kill chemotherapy-sensitive and -resistant human EOC cells and the likely mechanism of action contributing to GAEL-induced cell death, namely methuosis.
ResearchersWe postulate that induction of methuosis in chemotherapy-resistant EOC cells identifies a novel cell death pathway for the further development of new treatments for EOC patients.
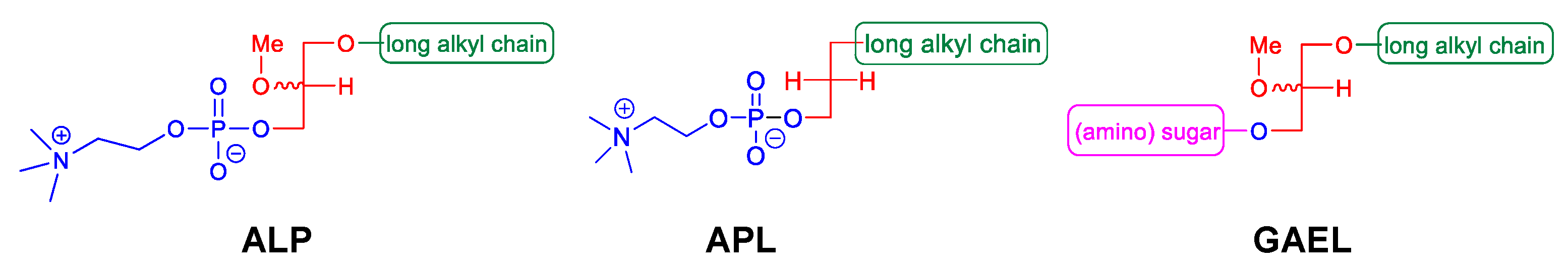
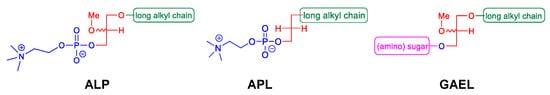
Figure 1.
Different classes of antitumor ether lipids (AELs); alkyllysophospholipids (ALP), alkylphospholipids (APL), and glycosylated antitumor ether lipids (GAEL).
2. GAELs: How Did We Get Here?
Antitumor ether lipids (AELs) are broadly defined as a group of synthetic ether lipid analogs with antitumor activity. They are comprised of three subclasses including alkyllysophospholipids (ALPs), alkylphospholipids (APLs), and GAELs (
Figure 1). The discovery of the antitumor activity of ether lipids originated from studies at the Max-Planck-Institut für Immunobiologie in Freiburg, Germany, in the 1960s. LPC, which is generated from hydrolysis of phosphatidylcholine by phospholipase A2, was found to strongly potentiate the phagocytic activity of peritoneal macrophages in vitro and in vivo
[53][54][55][53,54,55]. LPC is not a viable drug because it is rapidly metabolized and inactivated in cells by acyltransferases and lysophospholipases. To overcome this, metabolically stable LPC analogs were synthesized by replacing the acyl group at the sn-1 position with an alkyl group with 12–20 carbons. The sn-2 position was either an OH, H, or a methoxy (OCH
3) group
[56][57][58][56,57,58]. These ether LPC analogs, the ALPs (
Figure 1), demonstrated increased half-life and significantly higher ability to boost antibody production in response to various antigens compared to LPC
[59]. Thus, ALPs were initially developed for use as potential immunomodulators. Subsequently, a number of other ether LPC analogs were observed to have cytotoxic activity against a variety of cancer cells in vitro with 1-O-octadecyl-2-O-methyl-glycerophosphocholine (ET-18-OCH
3; aka edelfosine) being the most active
[60][61][62][60,61,62]. Further testing of ALPs using in vivo mouse and rat tumor models demonstrated antitumor activity by reducing tumor growth and metastasis
[63][64][63,64]. While several different pathways contributing to cell death in vitro have been examined, including inhibition of protein kinase C
[65][66][65,66], decreasing arachidonate release
[66], abrogating phosphatidylcholine biosynthesis
[67][68][67,68], and altering cell membranes
[69][70][69,70], edelfosine exhibited antitumor activity against a wide range of cancer cells by inducing apoptosis
[71][72][71,72]. ALPs have been tested in clinical trials and exhibit high tolerance to reversible toxicities and produce tumor stasis or a decrease in the tumor growth rate
[63][64][71][73][74][75][76][77][63,64,71,73,74,75,76,77]. Despite having promising clinical efficacy in vivo, the compounds are not indicated for the treatment of human cancers.
Studies to identify the minimum structure of ALPs required for anticancer activity led to the development of the alkylphospholipids (APLs;
Figure 1). These compounds lack the glycerol backbone and consist of an alkylchain esterified to a phosphobase. The prototype of this group is hexadecylphosphocholine (miltefosine)
[71][78][71,78]. While miltefosine was initially investigated as an anticancer agent, tests were stopped due to toxic side effects. It is currently used for leishmaniasis and amoeboid infections. Several analogs of APLs, including erucylphosphocholine
[79] and perifosine
[80][81][80,81], have been synthesized and their antitumor activities characterized
[82]. Perifosine was under development to treat human cancers as it blocks PI3K/AKT/mTOR signaling
[83]. Perifosine showed considerable promise in early phase clinical trials but failed in Phase III trials for colorectal cancer and refractory multiple myeloma
[84]. The mode of action of these compounds appears to be similar to that of the ALPs in killing cells by inducing apoptosis
[79][82][79,82].
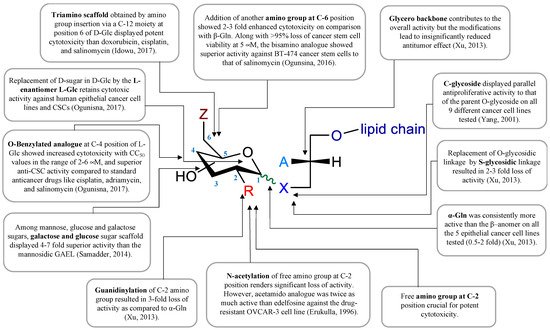
GAELs were developed by replacing the phosphocholine of edelfosine with a monosaccharide moiety via an O-glycosidic bond
[85]. The cancer cell killing activity of the GAEL compounds that were initially synthesized was weak compared to that of edelfosine. Substitution of the O-glycosidic to an S-glycosidic bond to improve lipophilicity did not significantly change the cancer cell-killing characteristics of the GAEL compounds
[86][87][86,87]. Subsequent structure–activity relationship (SAR) studies determined that replacing the OH at position 2 of the glucose with an NH
2 yielded cancer cell-killing activities that were similar or superior to edelfosine
[88]. This discovery opened up the field and led to the development of potent analogs and the further identification and characterization that GAELs were distinct from ALPs and APLs by inducing a non-apoptotic (caspase-independent) form of cell death.
3. GAEL Structure–Activity Relationship
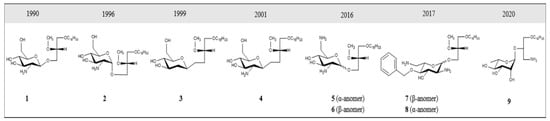
Several GAEL prototypes have been described
[50][52][88][89][90][91][92][93][50,52,88,89,90,91,92,93] (
Figure 2). The key event to the development of these compounds was the discovery by the Bittman group that the replacement of neutral thio-glucose by a basic 2-amino-β-D-glucose scaffold produced a potent GAEL [β-GLN (1-O-hexadecyl-2-O-methyl-3-O-(2′-amino-2′-deoxy-β-D-glucopyranosyl)-sn-glycerol)] with antiproliferative effects (
Figure 2, compound
2). Cell killing activity was tested in various human epithelial cancer cell lines, including A549, MCF-7, A427, and T84, with the greatest sensitivity in HGSOC OVCAR-3 cells
[88]. Indeed, compared to edelfosine, β-GLN was five times more effective at killing OVCAR-3 cells
[88]. Further experiments showed that acylation of the basic 2-amino group in β-GLN resulted in a 2–3-fold reduction in anticancer activity, indicating that the basic primary 2-amino group is critical for the antitumor effect. Further modification of β-GLN to contain an α-glucosidic linkage of 2-amino-D-glucose (α-GLN;
Figure 2, compound
1) displayed two-fold enhanced cell-killing activity compared to β-GLN
[91]. By contrast, replacement of the O-glycosidic linkage with S-glycosidic linkage and modifications on the glycero-ether moiety, in addition to guanidinylation of the amino group, resulted in reduced cell-killing effects
[91]. In order to increase the metabolic stability of β-GLN, C-glycosidic analogs were generated (
Figure 2, compounds
3 and
4) that retained antiproliferative effects
[92][93][92,93]. Studies to replace the α-O-glucosidic linkage with an α-O-galactosidic or α-O-mannosidic linkage revealed that both α-O-galactosidic and α-O-glucosidic linkages displayed comparable cell-killing effects while a >4-fold reduction in cell killing activity was observed with the α-O-mannosylated analog
[52]. Introducing a second amino group at the 6-position in α-GLN or β-GLN (
Figure 2, compounds
5 and
6) enhanced the cell-killing effect. Interestingly, both dibasic analogs displayed potent anticancer stem cell (CSC) activity against BT-474 and DU-145 stem cells with superior activity compared to salinomycin
[50]. Dibasic analogs of α-GLN or β-GLN were produced in which
D-glucose (
D-Glc) was replaced by glycosidase-resistant
L-glucose (
L-Glc) (
Figure 2, compounds
7 and
8)
[90]. Moreover, decoration of the dibasic
L-Glc scaffold by attachment of a phenyl group further enhanced the cell-killing effect
[90]. The incorporation of a third amino group and amphiphilic modulation of the
D-Glc scaffold also enhanced the cell-killing effects of GAELs
[94]. While first-generation GAELs were synthesized with
D-linked-sugars, their potential susceptibility to circulating and cellular glycosidases in vivo led to the development of
L-sugar-linked GAEL analogs that demonstrated equivalent or enhanced activity
[90]. Humans do not metabolize
L-sugars; thus,
L-GAELs are predicted to have longer half-lives in vivo. A structure–activity map that summarizes the structural modifications of the antitumor effect is shown in
Figure 3.
Figure 2. Structural evolution of GAEL prototypes.
1 = α-GLN,
2 = β-GLN;
3 and
4 are C-glycosidic analogs;
5 and
6 incorporate a second amino group at the 6 position; dibasic analogs
7 and
8 with
L-glucose;
9 = 3-amino-1-O-hexadecyloxy-2R-(O–α-
L-Rhamnopyranosyl)-sn-glycerol (
L-Rham).
Figure 3. GAEL structure–activity relationship map. Data compiled from references [50][52][88][90][91][92][94]. GAEL structure–activity relationship map. Data compiled from references [50,52,88,90,91,92,94].
One of the challenges to exploiting GAELs for therapeutic applications and animal studies is the requirement for multi-step synthesis resulting in low isolation yields. Synthetic processes required 12 or more synthetic steps. To reduce the number of synthetic steps, a more easily accessible
L-rhamnose-based cationic lipid was generated (
Figure 2, compound
9; 3-amino-1-
O-hexadecyloxy-2R-(
O–α-
L-rhamnopyranosyl)-
sn-glycerol (
L-Rham))
[95]. This
L-Rham-based GAEL differs from previous GAELs by the glycosidic linkage and glycero backbone; this compound retained cell-killing effects and was found to possess antitumor effects and therefore was used for further in vitro and in vivo studies
[49][95][49,95]. GAELs possess many desirable anticancer characteristics that include: (1) killing chemoresistant cell lines and primary HGSOC cells grown as 2D or 3D cultures and causing the disaggregation of 3D spheroids
[83][84][92][93][83,84,92,93]; (2) killing human cancer stem cell-enriched fractions from breast and prostate cell lines
[83][84][94][83,84,94]; and (3) killing cells via an apoptosis-independent mechanism of action
[47][48][50][51][52][47,48,50,51,52]. This latter characteristic distinguishes GAELs from most of the currently used chemotherapeutic drugs that rely on apoptosis to kill HGSOC cells.