 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jinlong Li | -- | 3378 | 2022-08-19 10:39:33 | | | |
| 2 | Beatrix Zheng | + 16 word(s) | 3394 | 2022-08-19 11:24:01 | | | | |
| 3 | Beatrix Zheng | -1 word(s) | 3393 | 2022-08-25 10:09:34 | | |
Video Upload Options
As a critical molecule in the onset and sustainment of inflammatory response, the receptor for advanced glycation end products (RAGE) has a variety of ligands, such as advanced glycation end products (AGEs), S100/calcium granule protein, and high-mobility group protein 1 (HMGB1). An increasing number studies have shown that RAGE ligand binding can initiate the intracellular signal cascade, affect intracellular signal transduction, stimulate the release of cytokines, and play a vital role in the occurrence and development of immune-related diseases, such as systemic lupus erythematosus, rheumatoid arthritis, and Alzheimer’s disease. In addition, other RAGE signaling pathways can play crucial roles in life activities, such as inflammation, apoptosis, autophagy, and endoplasmic reticulum stress. Therefore, the strategy of targeted intervention in the RAGE signaling pathway may have significant therapeutic potential, attracting increasing attention.
1. RAGE Structure
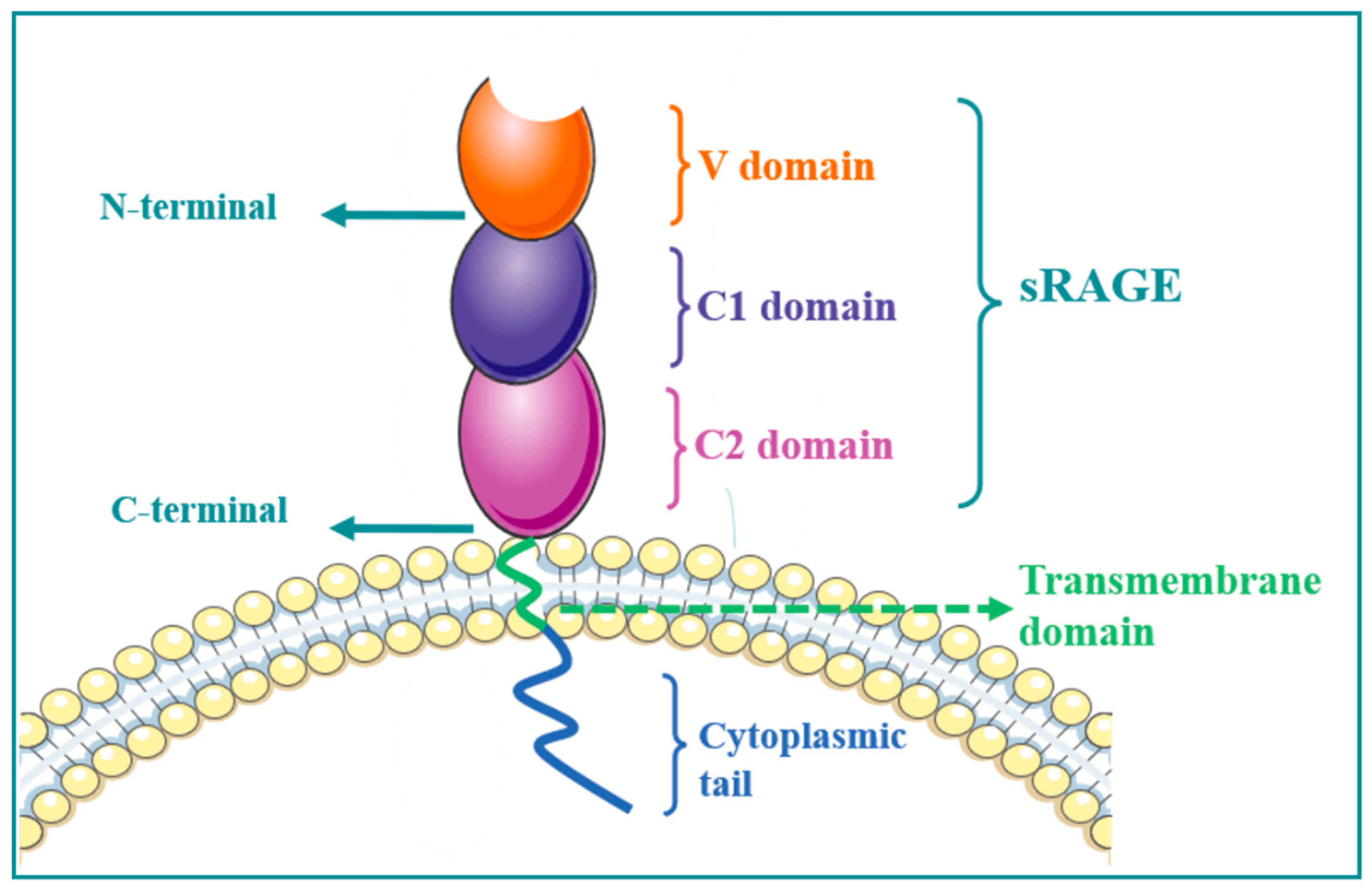
2. RAGE Regulates MAPK/NF-κB Signaling Pathway and Its Role in Immune-Associated Disease
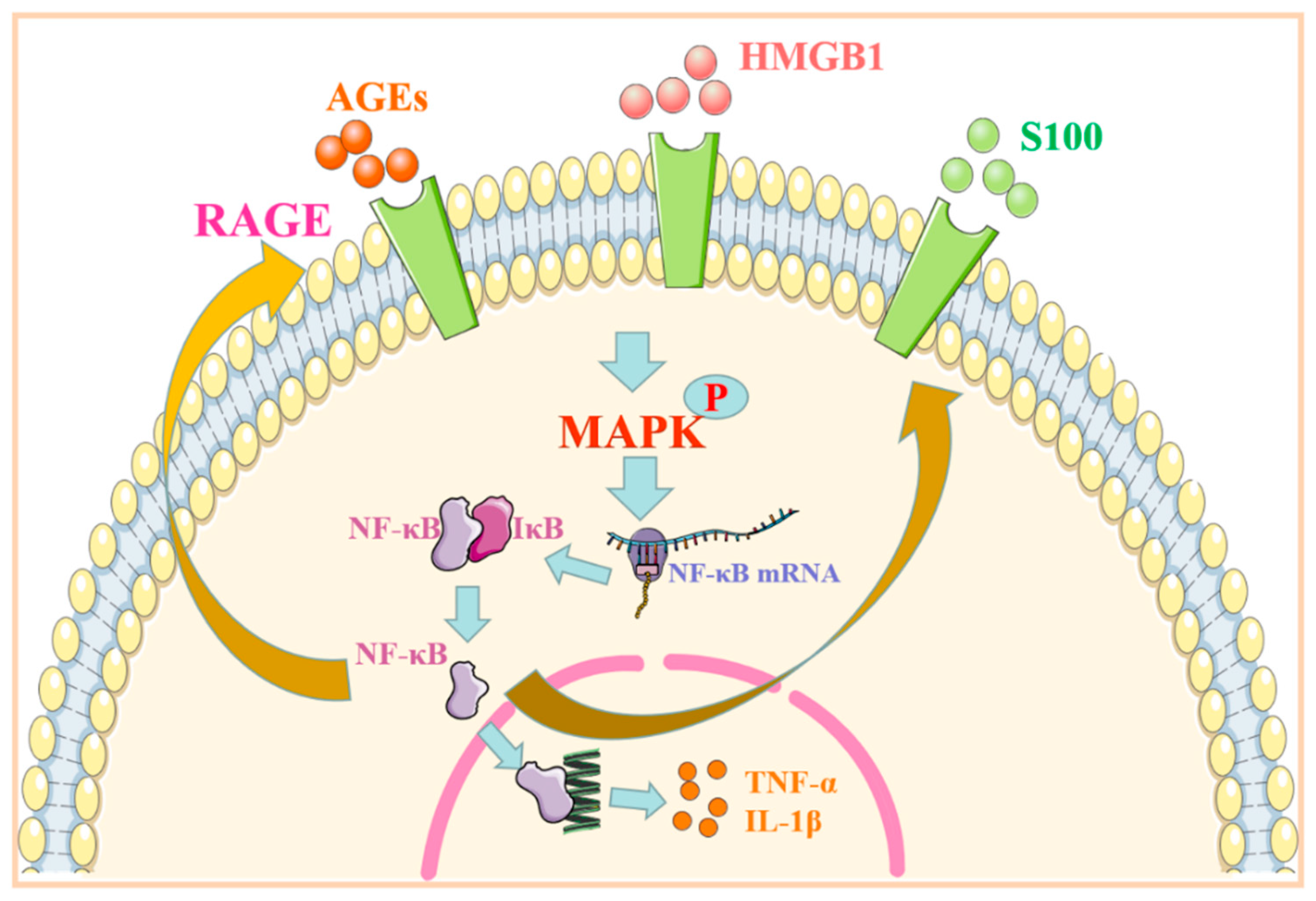
| Signal Pathway | Ligand | Specificity | Reaction | Diseases | References |
|---|---|---|---|---|---|
| HMGB1/RAGE-MAPK/NF-κB | HMGB1, AGEs | Endothelial cells | Production of inflammatory and B-cell-activating factors ↑ | SLE | [7] |
| Immune cells | Inflammatory reaction ↑ | [8] | |||
| Synovial cells | Inflammatory factor ↑ | RA | [9] | ||
| S100/RAGE-MAPK/NF-κB | S100A12 | Epithelial cells | EMT ↑ | Pulmonary fibrosis | [10] |
| S100A9 | Fibroblasts | Cell proliferation and secretion of inflammatory factors ↑ | [11] | ||
| Aβ/RAGE-MAPK/NF-κB | Aβ | Neurons | NFTs ↑ Cell activity ↓ Inflammatory factor ↑ |
AD | [12][13][14] |
| Aβ/RAGE-ERS | S100B | BBB | ERS ↑ Permeability of blood–brain barrier ↑ |
AD | [15][16][17] |
| S100B/RAGE-MEK/ERK1/2 | Neurons | Low concentrations of S100B protect nerve cells; very low and high concentrations of S100B produce neurotoxicity |
[18][19] |
2.1. RAGE Regulates MAPK/NF-kB Signaling Pathway and Its Role in Mediating Systemic Lupus Erythematosus
2.2. The Role of the MAPK/NF-kB Signaling Pathway Regulated by RAGE in Mediating Rheumatoid Arthritis
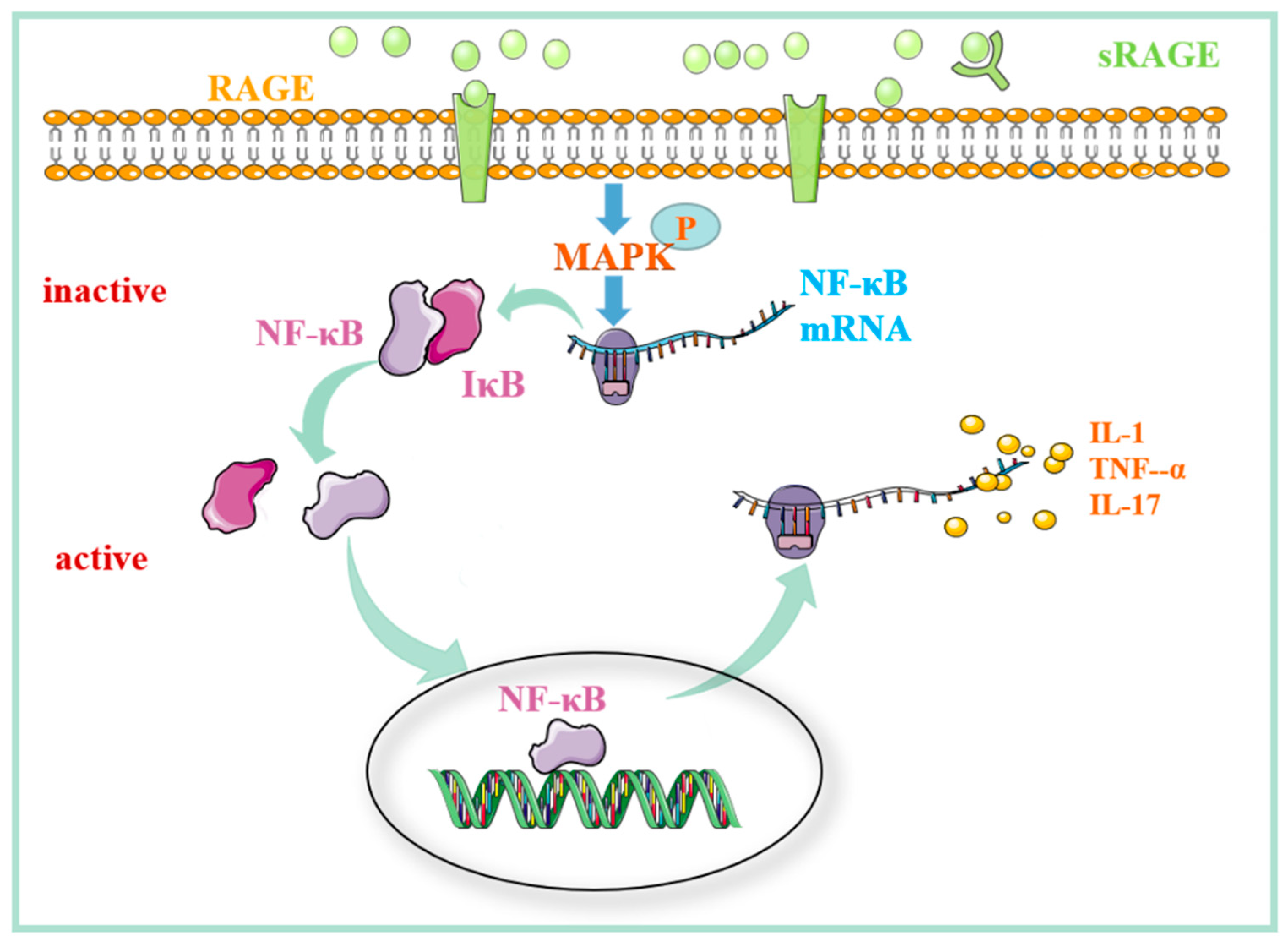
2.3. The Role of the MAPK/NF-kB Signaling Pathway Regulated by RAGE in Pulmonary Fibrosis
3. The Role of RAGE-Related Signaling Pathways in Alzheimer’s Disease
3.1. β-Amyloid Protein Regulates the RAGE-Related Signaling Pathway and Its Role in Alzheimer’s Disease
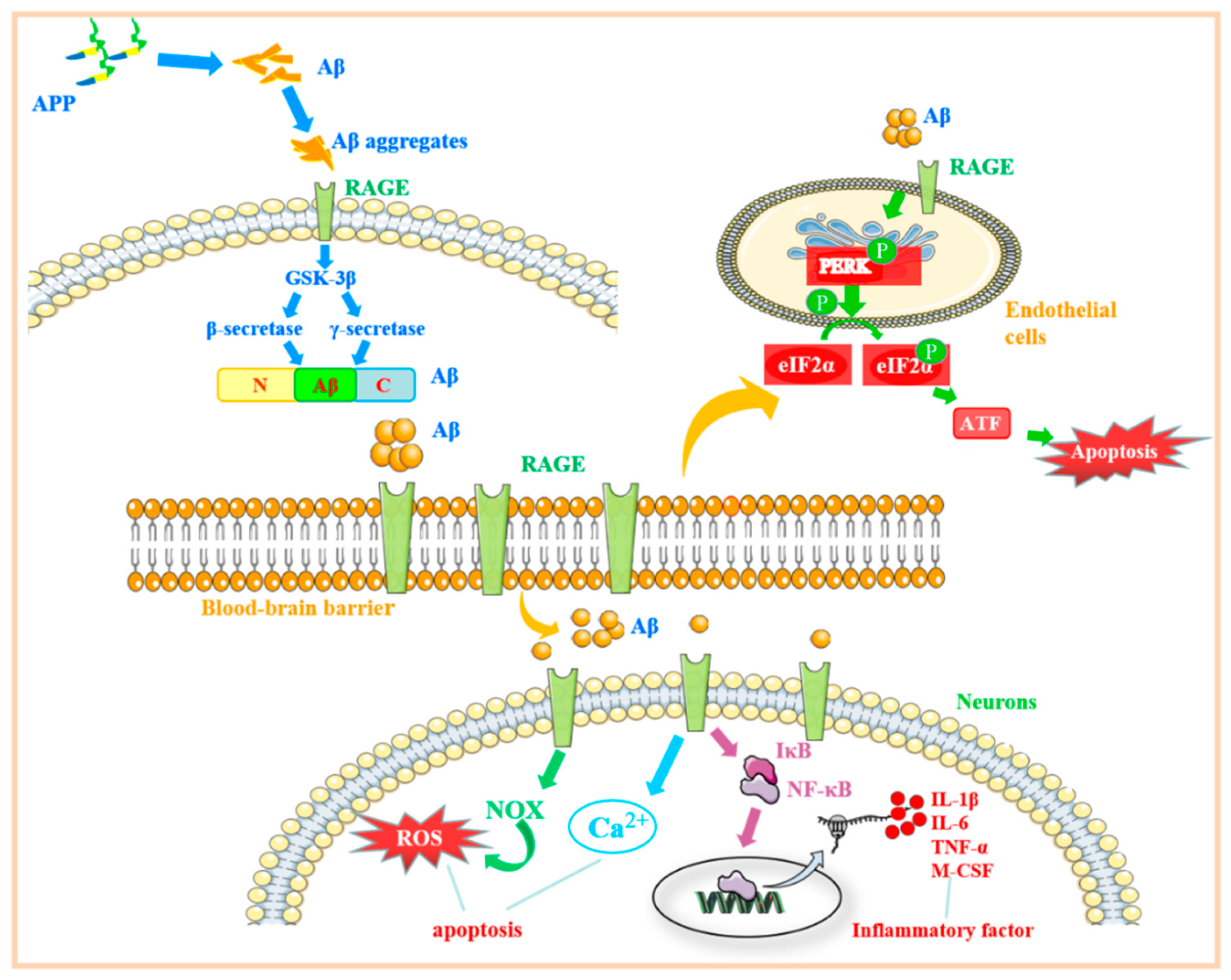
3.2. RAGE Regulates the Mechanisms Associated with Endoplasmic Reticulum Stress and Its Role in Alzheimer’s Disease
3.3. Regulation of RAGE-Related Signaling Pathway by S100B Protein and Its Role in Alzheimer’s Disease
3.4. The Mechanism of the RAGE-Related Pathway in AD
References
- Oh, S.; Son, M.; Choi, J.; Lee, S.; Byun, K. sRAGE prolonged stem cell survival and suppressed RAGE-related inflammatory cell and T lymphocyte accumulations in an Alzheimer’s disease model. Biochem. Biophys. Res. Commun. 2018, 495, 807–813.
- Li, S.T.; Dai, Q.; Zhang, S.X.; Liu, Y.J.; Yu, Q.Q.; Tan, F.; Lu, S.H.; Wang, Q.; Chen, J.W.; Huang, H.Q.; et al. Ulinastatin attenuates LPS-induced inflammation in mouse macrophage RAW264.7 cells by inhibiting the JNK/NF-kappaB signaling pathway and activating the PI3K/Akt/Nrf2 pathway. Acta Pharm. Sin. 2018, 39, 1294–1304.
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364.
- Ahmad, S.; Khan, M.Y.; Rafi, Z.; Khan, H.; Siddiqui, Z.; Rehman, S.; Shahab, U.; Khan, M.S.; Saeed, M.; Alouffi, S.; et al. Oxidation, glycation and glycoxidation-The vicious cycle and lung cancer. Semin. Cancer Biol. 2018, 49, 29–36.
- Laczko, R.; Chang, A.; Watanabe, L.; Petelo, M.; Kahaleua, K.; Bingham, J.P.; Csiszar, K. Anti-inflammatory activities of Waltheria indica extracts by modulating expression of IL-1B, TNF-alpha, TNFRII and NF-kappaB in human macrophages. Inflammopharmacology 2020, 28, 525–540.
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H491–H498.
- Maillard-Lefebvre, H.; Boulanger, E.; Daroux, M.; Gaxatte, C.; Hudson, B.I.; Lambert, M. Soluble receptor for advanced glycation end products: A new biomarker in diagnosis and prognosis of chronic inflammatory diseases. Rheumatology 2009, 48, 1190–1196.
- Martens, H.A.; Nienhuis, H.L.; Gross, S.; van der Steege, G.; Brouwer, E.; Berden, J.H.; de Sevaux, R.G.; Derksen, R.H.; Voskuyl, A.E.; Berger, S.P.; et al. Receptor for advanced glycation end products (RAGE) polymorphisms are associated with systemic lupus erythematosus and disease severity in lupus nephritis. Lupus 2012, 21, 959–968.
- Cerezo, L.A.; Remakova, M.; Tomcik, M.; Gay, S.; Neidhart, M.; Lukanidin, E.; Pavelka, K.; Grigorian, M.; Vencovsky, J.; Senolt, L. The metastasis-associated protein S100A4 promotes the inflammatory response of mononuclear cells via the TLR4 signalling pathway in rheumatoid arthritis. Rheumatology 2014, 53, 1520–1526.
- Lu, C.; Liu, J.; Yao, M.; Li, L.; Li, G. Downregulation of S100 calcium binding protein A12 inhibits the growth of glioma cells. BMC Cancer 2020, 20, 261.
- Xu, X.; Chen, H.; Zhu, X.; Ma, Y.; Liu, Q.; Xue, Y.; Chu, H.; Wu, W.; Wang, J.; Zou, H. S100A9 promotes human lung fibroblast cells activation through receptor for advanced glycation end-product-mediated extracellular-regulated kinase 1/2, mitogen-activated protein-kinase and nuclear factor-kappaB-dependent pathways. Clin. Exp. Immunol. 2013, 173, 523–535.
- Chakraborty, S.; ThimmaReddygari, J.; Selvaraj, D. G-Lymphatic, Vascular and Immune Pathways for Abeta Clearance Cascade and Therapeutic Targets For Alzheimer’s Disease. Comb. Chem. High Throughput Screen 2021, 24, 1083–1092.
- Da Costa Dias, B.; Jovanovic, K.; Gonsalves, D.; Weiss, S.F. Structural and mechanistic commonalities of amyloid-beta and the prion protein. Prion 2011, 5, 126–137.
- Cuevas, E.; Lantz, S.M.; Tobon-Velasco, J.C.; Newport, G.D.; Wu, Q.; Virmani, A.; Ali, S.F.; Santamaria, A. On the in vivo early toxic properties of A-beta 25–35 peptide in the rat hippocampus: Involvement of the Receptor-for-Advanced Glycation-End-Products and changes in gene expression. Neurotoxicol. Teratol. 2011, 33, 288–296.
- Chen, W.; Chan, Y.; Wan, W.; Li, Y.; Zhang, C. Abeta1–42 induces cell damage via RAGE-dependent endoplasmic reticulum stress in bEnd.3 cells. Exp. Cell Res. 2018, 362, 83–89.
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244.
- Bettigole, S.E.; Glimcher, L.H. Endoplasmic reticulum stress in immunity. Annu. Rev. Immunol. 2015, 33, 107–138.
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276.
- Prasad, K. AGE-RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell Biochem. 2019, 459, 95–112.
- Narvaez, J. Systemic lupus erythematosus 2020. Med. Clin. 2020, 155, 494–501.
- Wang, X.; Xiang, L.; Li, H.; Chen, P.; Feng, Y.; Zhang, J.; Yang, N.; Li, F.; Wang, Y.; Zhang, Q.; et al. The Role of HMGB1 Signaling Pathway in the Development and Progression of Hepatocellular Carcinoma: A Review. Int. J. Mol. Sci. 2015, 16, 22527–22540.
- Pisetsky, D.S. The complex role of DNA, histones and HMGB1 in the pathogenesis of SLE. Autoimmunity 2014, 47, 487–493.
- Li, J.; Xie, H.; Wen, T.; Liu, H.; Zhu, W.; Chen, X. Expression of high mobility group box chromosomal protein 1 and its modulating effects on downstream cytokines in systemic lupus erythematosus. J. Rheumatol. 2010, 37, 766–775.
- Yan, L.; Liang, M.; Yang, T.; Ji, J.; Jose Kumar Sreena, G.S.; Hou, X.; Cao, M.; Feng, Z. The Immunoregulatory Role of Myeloid-Derived Suppressor Cells in the Pathogenesis of Rheumatoid Arthritis. Front. Immunol. 2020, 11, 568362.
- Park, S.Y.; Lee, S.W.; Kim, H.Y.; Lee, W.S.; Hong, K.W.; Kim, C.D. HMGB1 induces angiogenesis in rheumatoid arthritis via HIF-1alpha activation. Eur. J. Immunol. 2015, 45, 1216–1227.
- Rojas, A.; Delgado-Lopez, F.; Perez-Castro, R.; Gonzalez, I.; Romero, J.; Rojas, I.; Araya, P.; Anazco, C.; Morales, E.; Llanos, J. HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumour Biol. 2016, 37, 3321–3329.
- Pullerits, R.; Bokarewa, M.; Dahlberg, L.; Tarkowski, A. Decreased levels of soluble receptor for advanced glycation end products in patients with rheumatoid arthritis indicating deficient inflammatory control. Arthritis Res. Ther. 2005, 7, R817–R824.
- Chen, Y.S.; Yan, W.; Geczy, C.L.; Brown, M.A.; Thomas, R. Serum levels of soluble receptor for advanced glycation end products and of S100 proteins are associated with inflammatory, autoantibody, and classical risk markers of joint and vascular damage in rheumatoid arthritis. Arthritis Res. Ther. 2009, 11, R39.
- Myles, A.; Viswanath, V.; Singh, Y.P.; Aggarwal, A. Soluble receptor for advanced glycation endproducts is decreased in patients with juvenile idiopathic arthritis (ERA category) and inversely correlates with disease activity and S100A12 levels. J. Rheumatol. 2011, 38, 1994–1999.
- Knani, I.; Bouzidi, H.; Zrour, S.; Bergaoui, N.; Hammami, M.; Kerkeni, M. Methylglyoxal: A Relevant Marker of Disease Activity in Patients with Rheumatoid Arthritis. Dis. Markers 2018, 2018, 8735926.
- Jafari Nakhjavani, M.R.; Jafarpour, M.; Ghorbanihaghjo, A.; Abedi Azar, S.; Malek Mahdavi, A. Relationship between serum-soluble receptor for advanced glycation end products (sRAGE) and disease activity in rheumatoid arthritis patients. Mod. Rheumatol. 2019, 29, 943–948.
- Inui, N.; Sakai, S.; Kitagawa, M. Molecular Pathogenesis of Pulmonary Fibrosis, with Focus on Pathways Related to TGF-beta and the Ubiquitin-Proteasome Pathway. Int. J. Mol. Sci. 2021, 22, 6107.
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712.
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863.
- Yamaguchi, K.; Iwamoto, H.; Horimasu, Y.; Ohshimo, S.; Fujitaka, K.; Hamada, H.; Mazur, W.; Kohno, N.; Hattori, N. AGER gene polymorphisms and soluble receptor for advanced glycation end product in patients with idiopathic pulmonary fibrosis. Respirology 2017, 22, 965–971.
- Grauen Larsen, H.; Marinkovic, G.; Nilsson, P.M.; Nilsson, J.; Engstrom, G.; Melander, O.; Orho-Melander, M.; Schiopu, A. High Plasma sRAGE (Soluble Receptor for Advanced Glycation End Products) Is Associated With Slower Carotid Intima-Media Thickness Progression and Lower Risk for First-Time Coronary Events and Mortality. Arter. Thromb. Vasc. Biol. 2019, 39, 925–933.
- Wittkowski, H.; Sturrock, A.; van Zoelen, M.A.; Viemann, D.; van der Poll, T.; Hoidal, J.R.; Roth, J.; Foell, D. Neutrophil-derived S100A12 in acute lung injury and respiratory distress syndrome. Crit. Care Med. 2007, 35, 1369–1375.
- Mulrennan, S.; Baltic, S.; Aggarwal, S.; Wood, J.; Miranda, A.; Frost, F.; Kaye, J.; Thompson, P.J. The role of receptor for advanced glycation end products in airway inflammation in CF and CF related diabetes. Sci. Rep. 2015, 5, 8931.
- Ray, B.; Maloney, B.; Sambamurti, K.; Karnati, H.K.; Nelson, P.T.; Greig, N.H.; Lahiri, D.K. Rivastigmine modifies the alpha-secretase pathway and potentially early Alzheimer’s disease. Transl. Psychiatry 2020, 10, 47.
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193.
- Nan, K.; Han, Y.; Fang, Q.; Huang, C.; Yu, L.; Ge, W.; Xiang, F.; Tao, Y.X.; Cao, H.; Li, J. HMGB1 gene silencing inhibits neuroinflammation via down-regulation of NF-kappaB signaling in primary hippocampal neurons induced by Abeta25–35. Int. Immunopharmacol. 2019, 67, 294–301.
- Esposito, G.; Scuderi, C.; Lu, J.; Savani, C.; De Filippis, D.; Iuvone, T.; Steardo, L., Jr.; Sheen, V.; Steardo, L. S100B induces tau protein hyperphosphorylation via Dickopff-1 up-regulation and disrupts the Wnt pathway in human neural stem cells. J. Cell Mol. Med. 2008, 12, 914–927.
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913.
- Huang, Q.; Yang, Z.; Zhou, J.P.; Luo, Y. HMGB1 induces endothelial progenitor cells apoptosis via RAGE-dependent PERK/eIF2alpha pathway. Mol. Cell Biochem. 2017, 431, 67–74.
- Businaro, R.; Leone, S.; Fabrizi, C.; Sorci, G.; Donato, R.; Lauro, G.M.; Fumagalli, L. S100B protects LAN-5 neuroblastoma cells against Abeta amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. J. Neurosci. Res. 2006, 83, 897–906.

