1. RAGE Structure
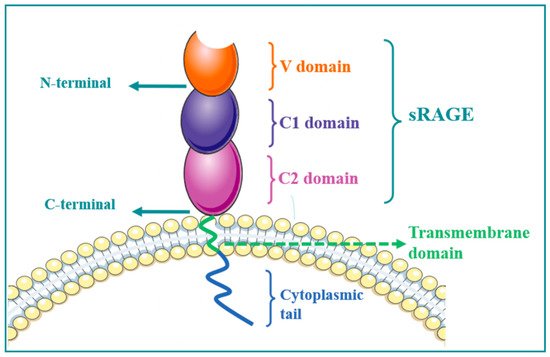
RAGE consists of extracellular, hydrophobic, transmembrane, and intracellular segments. The extracellular V-shaped region provides RAGE–ligand binding sites. Intracellular fragments can bind to various intracellular signal molecules and mediate signal transduction to cause cascade reactions, including full-length type, truncated C-terminal type, and truncated N-terminal type. The truncated C-terminal type is endogenous secretory soluble RAGE (esRAGE), which can be secreted by cells and contains only the extracellular segment. In contrast, the truncated N-terminal type consists of the transmembrane region and an intracellular component. Membrane-associated proteases can remove the transmembrane component of RAGE by hydrolysis, and the released extracellular segment can form soluble sRAGE with esRAGE. sRAGE can competitively bind to RAGE ligands, but binding to ligands terminates intracellular signal transduction due to the loss of transmembrane and intracellular fragments [
14] (
Figure 1).
Figure 1. RAGE structural organization. The extracellular domain comprises three domains: V, C1, and C2. One transmembrane receptor passes through the plasma membrane bilayer, followed by an intracellular cytoplasmic tail. The extracellular region without a transmembrane receptor and a cytoplasmic tail is called soluble RAGE (sRAGE).
2. RAGE Regulates MAPK/NF-κB Signaling Pathway and Its Role in Immune-Associated Disease
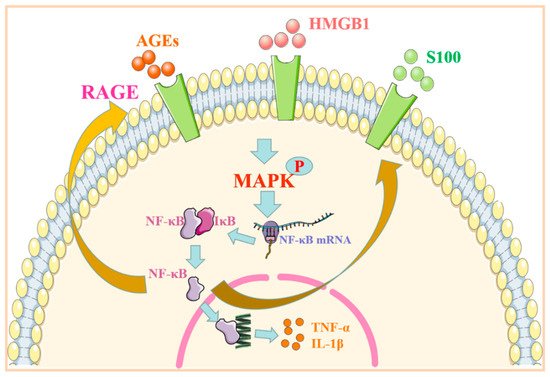
As an essential protein complex, NF-κB can be found in almost all animal cells. It can affect cell growth, differentiation, and apoptosis by controlling critical physiological processes, such as DNA transcription and cytokine production. In resting cells, NF-κB usually binds to its protein inhibitor of NF-κB (IκB). Therefore, it is in a state of inhibition and does not exert the function of transcriptional activity. After activation, the MAPK pathway can hydrolyze IκB and dissociate it from NF-κB. The free NF-κB enters the nucleus to regulate the transcriptional expression of inflammatory factors [
33]. The face of RAGE on the cell membrane is at a low level under normal physiological conditions. When the body’s inflammatory factors increase, the body is traumatized or suffers from abnormal conditions, such as diabetes. The content of ligands, such as AGEs and HMGB1, increase, and the expression of RAGE is upregulated [
1]. When RAGE is combined with ligands, it promotes the downstream phosphorylation of the p38 MAPK protein, thus increasing the expression of the NF-κB signaling path [
34]. NF-κB encourages the expression of inflammatory factors, such as TNF-α and IL-1β, by regulating the target genes and triggering related inflammatory and autoimmune responses, resulting in persistent tissue damage [
35]. When NF-κB is activated, it can promote the binding of RAGE to its ligand and further encourage the continuous expression of cytokines and tissue factors [
36] (
Figure 2).
Figure 2. RAGE regulates the MAPK/NF-κB signaling pathway. After binding to the ligand, RAGE phosphorylates its downstream MAPK and activates NF-κB protein. NF-κB enters the nucleus to promote the transcriptional expression of inflammatory factors. Abbreviations: AGEs: advanced glycation end products; HMGB1: high-mobility group protein 1; RAGE: receptor for advanced glycation end products; MAPK: mitogen-activated protein kinase; NF-κB: nuclear factor kappa-B; TNF-α: tumor necrosis factor α; IL-1β: interleukin-1β.
Table 1 summarizes the related ligands and mechanisms of the RAGE-MAPK/NF-κB pathway in SLE, RA, pulmonary fibrosis, and AD.
Table 1. The role of RAGE-related signaling pathways in immune-associated diseases.
2.1. RAGE Regulates MAPK/NF-kB Signaling Pathway and Its Role in Mediating Systemic Lupus Erythematosus
SLE is an autoimmune disease involving various systems and organs, with diverse and variable clinical manifestations. The pathogenesis of SLE is complicated. The interaction between genetic and environmental factors, the production of pathogenic antibodies, and the deposition of immune complexes (ICs) formed by combining autoantibodies and antigens are all causes of SLE [
50]. HMGB1 binds to Toll-like receptors, such as TRL-2, TRL4, and RAGE, to regulate the immunoinflammatory response by mediating the MAPK/NF-κB signal pathway. In addition, it has been found that the immune complex formed by the combination of HMGB1 and DNA plays a vital role in the pathogenesis of SLE [
51,
52]. These findings suggest that HMGB1 plays a crucial role in the occurrence and development of SLE.
To confirm the above conjecture, some studies have further examined the level of HMGB1 in SLE patients and animal models. The results showed that the expression level of HMGB1 in patients and animals increased, and the inflammatory response of related immune cells increased significantly, which led to the occurrence of SLE [
37]. Immune complexes can activate the expression of RAGE in human endothelial cells, participate in the response of the HMGB1-RAGE axis in promoting SLE vasculitis, and induce the production of TNF-α and B-cell-activating factors. Some studies have revealed that plasma HMGB1 levels in patients with SLE are significantly higher than those in healthy subjects and positively correlated with the concentration of plasma antinuclear antibody [
53]. In addition, the concentration of AGEs in skin cells and serum, as well as the expression of RAGE mRNA in peripheral blood monocytes, are significantly increased in patients with SLE. Immunohistochemical detection revealed almost no expression of RAGE in normal glomerular cells of normal subjects. However, prominent expression still occurs in glomeruli patients with SLE [
38]. In summary, we can speculate that the possible mechanism of RAGE involves binding to ligands and activation of transcription factor NF-κB through the MAPK pathway to enhance inflammatory response and promote SLE. At present, direct evidence about the involvement of RAGE in SLE is rare, whereas the role of its ligands in SLE is more prominent; therefore, the mechanism of RAGE in SLE requires further investigation.
2.2. The Role of the MAPK/NF-kB Signaling Pathway Regulated by RAGE in Mediating Rheumatoid Arthritis
RA is characterized by inflammatory synovitis, leading to joint injury and deformity. Although the formation of RA is not precise, the overexpression of many cytokines is a significant cause of bone destruction. The cytokines that play a critical role in this process are interleukin-1β (IL-1β) and TNF-α [
54]. Previous studies have confirmed that HMGBl is highly expressed in the synovium of patients with RA, and anti-HMGB1 antibody can inhibit the inflammatory response caused by HMGB1, reduce the damage to joint tissue, and effectively alleviate arthritis caused by HMGB1 [
55]. As one of the ligands with the highest affinity for RAGE, HMGB1 activates NF-κB mainly through an RAS/MAPK kinase cascade reaction after binding to each other and induces the production of a large number of cytokines and inflammatory factors [
56].
Comparison of the expression of RAGE mRNA, TNF-α, and IL-1β in synovial tissue of patients with rheumatoid arthritis and osteoarthritis revealed that the concentration of TNF-α and IL-1β in synovial tissue of the former was higher than that of the latter. There was a significant positive correlation between the attention of two inflammatory factors and the expression of RAGE. These results suggest that HMGBl-RAGE can promote the expression of inflammatory cytokines by activating NF-κB [
39]. It can be inferred that HMGBl-RAGE upregulates the expression of inflammatory factors in RA by activating NF-κB. In addition, the test revealed that patients positive for serum rheumatoid factor had lower serum sRAGE levels than patients with RA disease without a rheumatoid factor [
57]; a number of studies have corroborated these results [
58,
59,
60]. Recent studies have revealed that compared with healthy controls, RA patients have higher serum sRAGE levels, which is positively correlated with disease activity [
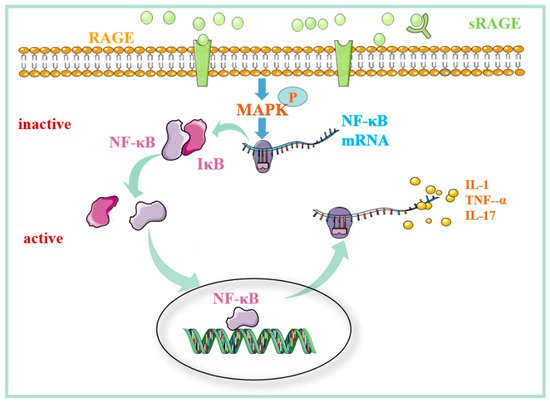
61]. This is phenomenon is contrary to the results reported in previous studies. We speculate that the increase in serum sRAGE levels is most likely due to the decrease in proinflammatory RAGE–ligand binding when disease activity is controlled. High levels of sRAGE production and secretion may indicate the response of compensatory anti-inflammatory mechanisms to tissue damage by acting as bait receptors and proinflammatory ligands and preventing them from transmitting harmful signals. This theory further supports the hypothesis that decreased sRAGE levels in RA patients may increase inflammatory tendencies. In summary, HMGB1/RAGE can damage joints by regulating the MAPK/NF-κB signal pathway, resulting in RA-related symptoms. Furthermore, by competitively binding RAGE ligands, sRAGE inhibits the entry of inflammatory cells into the joint cavity (
Figure 3).
Figure 3. The mechanism of action of RAGE–ligand binding in RA. The binding of RAGE with the ligand activates the MAPK/NF-κB signaling pathway and induces the production of inflammatory factors. sRAGE can block the ligand–RAGE interaction on the cell surface, reducing the entry of inflammatory cells into the joint cavity. Abbreviations: RAGE: receptor for advanced glycation end products; sRAGE: soluble receptor for advanced glycation end products; MAPK: mitogen-activated protein kinase; NF-κB: nuclear factor kappa-B; IκB: inhibitor of NF-κB; TNF-α: tumor necrosis factor α; IL-1: interleukin-1; IL-17: interleukin-17.
2.3. The Role of the MAPK/NF-kB Signaling Pathway Regulated by RAGE in Pulmonary Fibrosis
When the lungs are injured, immune cells (such as monocytes, macrophages, T cells, B cells, and NK cells) and some non-immune cells (such as endothelial cells, epidermal cells, and fibroblasts) secrete a variety of inflammatory, profibrotic cytokines and chemokines, leading to persistent inflammation in the lungs and accumulation of extracellular matrix, ultimately leading to pulmonary fibrosis disease. In the pathogenesis of idiopathic pulmonary fibrosis, it is indispensable for epithelial cells to form myofibroblasts through epithelial–mesenchymal transition (EMT) [
62]. RAGE was found to be significantly expressed on the membrane of alveolar macrophages, vascular smooth muscle cells, pulmonary endothelia, and epithelial cells [
63,
64]. Considerable evidence has shown that the interaction between RAGE and ligands is essential in pathological processes, such as inflammation and fibrosis [
65,
66]. S100A12 is reported to be highly expressed in the lungs of patients with acute respiratory distress syndrome, which activates pulmonary inflammation and endothelial cells through the binding of RAGE, leading to an increase in fibrogenic growth factor and the induction of epithelial–mesenchymal transformation [
40,
67]. A large amount of HMGB1 can induce EMT in normal alveolar type II epithelial cells, although it is difficult for alveolar type II epithelial cells without the RAGE genotype to produce EMT, suggesting that HMGB1–RAGE–ligand interaction can induce the EMT process of alveolar type II epithelial cells [
68]. Through the stimulation of pulmonary interstitial cells in vitro, it was found that S100A9 stimulation of fibroblasts can significantly increase the expression of inflammatory factors in a dose-dependent manner. Treatment with ERK1/2MAPK inhibitors, anti-RAGE antibodies, and NF-κB inhibitors significantly downregulates S100A9-induced fibroblast proliferation and inflammatory cytokine secretion. It was confirmed that S100A9 and RAGE can activate fibroblasts through ERK1/2, MAPK, and NF-κB signaling pathways [
41]. In summary, S100 protein RAGE can mediate the secretion of inflammatory factors and promote fibrosis through the MAPK pathway.
MAPK/NF-KB, as the first confirmed RAGE regulatory pathway, is also a classic inflammatory pathway. Whether AGEs, HMGB1, or S100 protein family members, RAGE classic ligands can activate the MAPK/NF-KB pathway and promote the occurrence and development of SLE, RA, pulmonary fibrosis, and other immune diseases.
3. The Role of RAGE-Related Signaling Pathways in Alzheimer’s Disease
3.1. β-Amyloid Protein Regulates the RAGE-Related Signaling Pathway and Its Role in Alzheimer’s Disease
AD is a neurodegenerative disease characterized by memory impairment, executive dysfunction, and personality and behavioral changes. In recent years, an increasing number of studies have found that AD is also an immune-related disease, and the inflammation caused by the brain immune system can drive the deterioration of AD. However, its pathogenesis has not been studied. The mainstream theory is that β-secretase and γ-secretase on the mass membrane are cut sequentially at the N and C ends of the amyloid prebiotic protein (APP) to form a β-amyloid peptide. The primary forms are Aβ40 and Aβ42 [
69]. There are three different soluble forms of Aβ in the brain: haplotype Aβ, Aβ aggregate, and filamentous Aβ [
70]. The proportion of Aβ42 content increases and accumulates in the patient’s brain to form neurofilaments and Aβ aggregates. Increasing evidence in recent years shows that Aβ aggregates are the “culprit” of this neurodegenerative disease. The deposited Aβ aggregates cause neurofibrillary tangles (NFTs) and senile plaques to form in the brain. In contrast, NFTs and senile plaques can reduce the activity of neurons and cause neurotoxic damage, leading to inflammation of the nervous system. Studies have shown that deposited Aβ aggregates can activate the expression of RAGE. Compared with non-AD patients, the expression of RAGE in hippocampal neurons, astrocytes, microglia, and endothelial cells was found to be significantly increased in AD patients [
42,
71]. In addition, a series of experiments have shown that Aβ can cause neurotoxicity in mice, inducing nerve apoptosis, which is enhanced by the coaction of Aβ and RAGE [
43].
In experiments, Cuevas and colleagues found that after injecting Aβ into the hippocampus of rats, the expression of RAGE increased [
44], and the downstream pathway protein NF-κB was also activated. Guglielmotto et al. found that two different AGEs can upregulate the expression of Aβ-secretase BACE1 by binding to RAGE through inhibitor treatment of neuroblastoma cells differentiated by SK-N-BE [
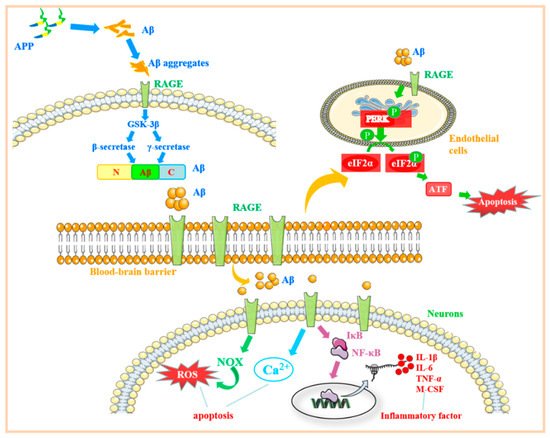
72]. In Neuro-2a cells transfected with RAGE-EGFP, RAGE-Aβ can activate signal transduction pathways, including p38MAPK, and activate NF-κB and AP-1 proteins and promote the production of inflammatory cytokines IL-6, TNF-α, and macrophage colony-stimulating factor (M-CSF), eventually leading to an inflammatory response. In summary, the combination of RAGE on the nerve cell membrane and Aβ can activate NF-κB to mediate the inflammatory response and play an essential role in developing Alzheimer’s disease (
Figure 4).
Figure 4. RAGE-related signaling pathways in Alzheimer’s disease. After APP is converted to Aβ, on the one hand, it combines with RAGE to activate the downstream MAPK/NF-κB pathway to release inflammatory factors and damages nerve cells. On the other hand, Aβ binds to RAGE on the surface of BBB cells, causing ERS, altering the permeability of the BBB, increasing the concentration of Aβ in the central nervous system, and aggravating AD-related symptoms. Abbreviations: APP: amyloid prebiotic protein; Aβ: amyloid β-protein; RAGE: receptor for advanced glycation end products; GSK-3β: glycogen synthase kinase 3β; PERK: protein kinase R-like endoplasmic reticulum kinase; eIF2α: eukaryotic translation initiation factor 2α; NOX: nitrogen oxide; NF-κB: nuclear factor kappa-B; IκB: inhibitor of NF-κB; TNF-α: tumor necrosis factor α; IL-1β: interleukin-1β; IL-6: interleukin-6; M-CSF: macrophage colony-stimulating factor.
3.2. RAGE Regulates the Mechanisms Associated with Endoplasmic Reticulum Stress and Its Role in Alzheimer’s Disease
Under normal circumstances, the blood–brain barrier (BBB) can play a protective role and prevent Aβ in the blood from entering the central nervous system [
45,
48]. When AD occurs, the expression of RAGE on cerebral microvessels increases significantly. After injecting Aβ into the blood of AD model mice, it was found that Aβ was transported to brain tissue in a process mediated by RAGE. Following anti-RAGE action, the transport process was blocked. Through an LG-RAGE particular antibody blocking test and ap radiotracer assay, it was ultimately confirmed that Aβ penetrated the BBB in a process mediated by RAGE, a specific receptor on the vascular wall [
73]. It was found that the potential mechanism of AD blood–brain barrier destruction is the activation of endoplasmic reticulum stress (ERS) in a dose-dependent manner [
46].
The endoplasmic reticulum is an important location of protein processing in cells and plays a vital role in the synthesis, folding, assembly, and modification of soluble proteins and membrane proteins. When exogenous harmful substances disrupt the normal folding reaction of proteins, many abnormal proteins accumulate in the endoplasmic reticulum, triggering ERS.
Some studies have revealed that the RAGE-related signaling pathway can activate the protein kinase R-like endoplasmic reticulum kinase (PERK) pathway in ERS to control the apoptosis of human endothelial progenitor cells and affect the pathogenesis of cardiovascular disease [
74]. PERK is a type I transmembrane protein that exists in the endoplasmic reticulum. It can phosphorylate the alpha subunit of eukaryotic translation initiation factor 2α (eIF2α) protein kinase. When unfolded or misfolded, proteins accumulate in the cell, and the immunoglobulin heavy-chain binding protein (BiP) is released from PERK and phosphorylates eIF2α. After phosphorylation, eIF2α can induce the expression of activating transcription factor 4 (ATF4), which activates the expression of unfolded protein response (UPR) target genes and promotes apoptosis [
47]. Chen also studied the potential mechanism of the destruction of the blood–brain barrier in Alzheimer’s disease and found that Aβ42-induced RAGE activates ERS in a dose-dependent manner [
45]. When cells were transfected with RAGE-siRNA, the endoplasmic reticulum stress markers decreased under the induction of Aβ42. In summary, Aβ can activate endothelial cell ERS through RAGE and increase the permeability of the blood–brain barrier. Therefore, the concentration of Aβ in the central nervous system increases, aggravating the AD condition (
Figure 4).
3.3. Regulation of RAGE-Related Signaling Pathway by S100B Protein and Its Role in Alzheimer’s Disease
S100B is an essential member of the S100 protein family and is highly expressed in astrocytes. Under the induction of some stimuli, astrocytes can release a certain amount of S100B [
49]. Through immunoblotting, EMSA ultrasonography, and reverse transcriptase-polymerase chain reaction techniques, experiments have proven that micromolar S100B can stimulate c-Jun N-terminal kinase (cJNK) phosphorylation by binding to RAGE and activating nuclear AP-1/cJun in cultured human neural stem cell transcription. In addition, Western blot, siRNA, and immunofluorescence analyses have shown that phosphorylated cJNK induced by S100B can promote the formation of NFTs in AD.
Recent studies have revealed that S100B can protect LAN-5 nerve cells at the nanomolecular level by upregulating antiapoptotic factor Bcl-2 and inhibiting the neurotoxic Aβ25–35 peptide, which can reduce the level of Bcl-2 [
75]. At this concentration, S100B binds to RAGE and activates the downstream MEK-ERK1/2 pathway to stimulate the expression of survival-related genes and produce an antiapoptosis effect. After adding the MEK inhibitor, the ability of S100B to upregulate the expression of Bcl-2 was inhibited, which further confirmed the above pathway. At high concentrations, for example, due to astrocyte death, damaged astrocyte leakage, and protein clearance defects, when the intercellular concentration of S100B is expected to be higher than that of 500 nM, S100B not only does not protect LAN-5 cells from neurotoxicity from Aβ 25–35 peptide but also exerts neurotoxicity itself, which could further increase the neurotoxicity of Aβ 25–35 peptide. Furthermore, S100B was found to overactivate RAGE and its downstream ERK1/2 pathway, causing oxidative stress and inducing neuronal death. In summary, varying concentrations of S100B combined with RAGE play an opposite role in AD by activating different signal pathways. A low concentration of S100B can protect nerve cells, whereas low and high concentrations of S100B result in neurotoxicity.
3.4. The Mechanism of the RAGE-Related Pathway in AD
In addition to its neurotoxicity, which can activate inflammatory factors in nerve cells through RAGE and induce nerve cell apoptosis, Aβ can also combine with RAGE on the surface of endothelial cells to cause endoplasmic reticulum stress, which pathologically increases the permeability of the blood–brain barrier, increases the concentration of Aβ in the central nervous system, and further aggravates the injury of nerve cells. In addition, a high concentration of S100B was found to produce certain neurotoxicity in combination with RAGE. The combination of Aβ and S100B with RAGE shows that RAGE plays an important role in the occurrence and development of AD.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27154922