Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Frederik De Smet | -- | 3331 | 2022-08-17 04:05:26 | | | |
| 2 | Catherine Yang | Meta information modification | 3331 | 2022-08-17 04:09:43 | | | | |
| 3 | Beatrix Zheng | Meta information modification | 3331 | 2022-08-23 05:11:08 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Panovska, D.; Smet, F.D. Functional Precision Oncology on Glioblastoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/26229 (accessed on 27 July 2026).
Panovska D, Smet FD. Functional Precision Oncology on Glioblastoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/26229. Accessed July 27, 2026.
Panovska, Dena, Frederik De Smet. "Functional Precision Oncology on Glioblastoma" Encyclopedia, https://encyclopedia.pub/entry/26229 (accessed July 27, 2026).
Panovska, D., & Smet, F.D. (2022, August 17). Functional Precision Oncology on Glioblastoma. In Encyclopedia. https://encyclopedia.pub/entry/26229
Panovska, Dena and Frederik De Smet. "Functional Precision Oncology on Glioblastoma." Encyclopedia. Web. 17 August, 2022.
Copy Citation
Glioblastoma remains the most malignant and intrinsically resistant brain tumour in adults. Precision oncology refers to the evaluation of treatment efficacies and vulnerabilities of (ex vivo) living tumor cells in a highly personalized way. Precision oncology aims at identifying effective therapeutic approaches based on properties (biomarkers) that are specific to each patient’s tumor.
functional precision oncology
glioblastoma
drug sensitivity
1. The Complexity of GBM
Glioblastoma (GBM) [1], still the most malignant primary brain cancer in adults [2], significantly suffers from the above described drawbacks. Already since 2005, the standard-of-care treatment of GBM includes a multidisciplinary approach combining surgery, ionizing radiation (RT) and chemotherapy. In spite of this aggressive approach, the median survival of GBM patients generally does not exceed 2 years [3]. This is caused by a combination of factors. (i) GBM is a highly infiltrative tumor, meaning that surgeons are commonly unable to resect the entire tumor, resulting in significant amounts of residual disease. In line with this, the extent of resection (EOR) has been identified as an important prognostic factor for GBM [4]. (ii) Targeting the residual tumor cells, primarily done by a combination of radiation therapy and temozolomide (TMZ), turns out to be extremely difficult: already in more than 50% of patients, progressive disease is radiologically observed even before finishing TMZ treatment (typically already within 3 months of therapy) [3]. This strongly suggests that large amounts of intrinsically unresponsive tumor cells were residing in the brain tissue even before starting therapy, which rapidly cause recurrence in GBM patients. Identifying more suitable and patient-tailored therapies that are accessible to the central nervous system (CNS) and are able to target a heterogeneous population of tumor cells therefore remains a major challenge in achieving clinical benefits.
To identify appropriate drug targets for GBM, large-scale sequencing programs were initiated to uncover disease causing genetic aberrations [5][6][7]. Over the past decade, several hundreds of GBM tumors have been sequenced within various consortia, uncovering complex and elaborate genetic alterations, including single nucleotide variants, focal or large chromosomal deletions and/or amplifications, and gene fusions [7]. For several of these genetic aberrations, drugs that target the affected cellular pathways have been developed, either in the context of GBM or other cancer types. Examples of such targets/pathways include receptor tyrosine kinases (e.g., EGFR, PDGFRA, VEGFR, MET) and various downstream intracellular signaling pathways (e.g., PI3K, AKT, mTOR, MEK/ERK) [8], hyperactive fusion proteins (e.g., TACC-FGFR and NTRK-fusions) [9], DNA repair (ATR/CHK1/CHK2, MDM2/4, PARP1, WEE) [10][11], and cell cycle regulation (CDK4/6) [5]. In spite of numerous clinical trials that were conducted to test the efficacy of these drugs against GBM, clinical results have been disappointing [2].
The failure of these trials could in part be explained by a lack of sufficiently precise selection procedures to enroll the appropriate patients that could actually benefit from the given therapy [12]. So far, such selection has primarily been based on bulk genetic analyses, where the presence of specific genetic aberrations was used as inclusion criteria for assigning appropriate therapy for each patient—an approach used for instance in the INSIGhT trial for GBM patients (NCT02977780). However, the complexity and interpatient heterogeneity of the genetic aberrations in GBM are so extensive that multiple interfering pathways are often simultaneously affected [5][6][7], making it largely unclear whether tumor cells of a particular patient would be responsive to a given therapy (even in the presence of the particular targetable mutation). On top of this, with the advent of single-cell sequencing methods, it turns out that the cellular composition of a GBM tumor is more complex than initially anticipated. Indeed, single-cell RNA sequencing (scRNAseq) studies showed that multiple of the TCGA-based tumor cell subtypes and a variety of stem cell-like states (i.e., neural progenitor-like, astrocyte-like, oligodendrocyte progenitor-like and mesenchymal-like) can be simultaneously present in a single GBM tumor [13][14][15] while containing multiple, often divergent genomic aberrations. Moreover, the various stem cell-like cellular states are plastic, meaning that they are interchangeable, a process that seems driven by stress factors caused by the environment of the cells. As such, stem cell-like cells are often more resistant to therapeutic perturbations. Therefore, instead of the initially anticipated subgrouping into 4 major subtypes [5][16], current insights suggest that GBM tumors harbor dozens of different tumor cell profiles, probably each requiring a specific therapeutic approach [1].
Finally, GBM tumor cells can also acquire de novo resistance upon therapy. Indeed, in an initially TMZ-responsive tumor cell population, resistance can easily be acquired by upregulating DNA-repairing enzymes such as MGMT or by inactivating the DNA mismatch repair (MMR) system, eventually leading to tumor recurrence [17]. At this point, a second surgical resection is often used as salvage therapy combined with other chemotherapeutic agents, such as lomustine/CCNU [18]. Additionally, in the recurrent setting, it would be highly beneficial to have better tools available to identify more suitable therapeutic options. All the above shows that identifying an appropriate therapy for each GBM patient, either in the newly diagnosed or recurrent setting, remains a daunting task.
2. Functional Diagnostics: Evolving from a Static to a Dynamic Interrogation of Cancer Cells’ Ability to Respond to Therapy
Major efforts are currently being put in matching specific (genetic) cancer features to drug responses [19]. However, in order to determine therapeutic efficacy across different patients and within a single tumor, as highlighted above, genetic information alone is often proven insufficient. Indeed, most studies only use baseline measurements in a ‘static’ setting (i.e., one snapshot prior to treatment), and intend to correlate the presence of specific genetic features to subsequent responsiveness to therapy. The simultaneous aberration of multiple cellular pathways, which can significantly interfere with each other, or for which multiple therapeutic options are sometimes available, make it difficult to predict the most suitable therapy. A functional interpretation [20] (e.g., what happens before and after cells are exposed to a certain therapy; what are the effects of the drug on the cellular state) on the other hand could provide dynamic, faster, more detailed and potentially longitudinal insights into the ability of cells to respond to therapy in a genotype agnostic way, although methods to do so remain difficult. In this light, approaches for assessing differential drug responses are gaining traction by which live tumor cells are ex vivo exposed to various therapeutic insults, while a chosen cellular response is carefully monitored—an approach coined functional diagnostics, functional oncology or functional precision medicine [20]. When monitoring the right features, such approach does not necessarily require a complete biological understanding, while still providing medically relevant insights (e.g., do cells respond to therapy or not, rather than why do they respond or not), allowing faster translation to a clinical setting.
Functional diagnostics is, however, not a novel approach. Such strategy has been widely applied in other biomedical domains, such as infectious diseases where antibiogram screens are used to select the most appropriate antibiotic in a patient-tailored way. Still, translating such functional diagnostic assays to a cancer setting requires further amelioration and validation in order to become medically applicable.
Overall, the goal of functional testing is to bring forward personalized medicine to patients diagnosed with complex disease entities, where treatment options are rather limited. In other words, functional tests ought to facilitate the matching of each patient to the most beneficial treatment. This being said, ex vivo drug exposure of freshly isolated tumor biopsies can directly inform on cell death, alterations in signaling networks, cellular phenotype and morphology or even tumor cell–tumor microenvironment (TME) crosstalk and adverse events in normal tissue. Certainly, the type of functional readout informing on tumor and non-malignant cellular vulnerabilities would largely depend on the mechanism of action of the given treatment. Typically, investigators rely on commonly available, FDA-approved therapies or drugs in clinical trials where dose-escalating studies where safety and tolerability of the therapy of interest has been already assessed and approved.
In the particular case of GBM, the functional screening method should not only be able to map each tumor in great detail—given GBM’s high degree of intra-tumoral heterogeneity [21]—it should also be able to track molecular responses to drug treatments. Currently, every patient diagnosed with GBM is profiled using a uniformed diagnostic procedure, consisting of MRI scans and “static” measurements of pathological trademark alterations, such as chromosomal rearrangements, mutational patterns and MGMT promotor methylation (Figure 1). Functional testing gives the opportunity to directly evaluate therapy efficacy, either in dissociated GBM samples or tumor tissue slices [22]. In order to predict tumor cell behavior in such a complex and dynamic system as the GBM/brain, one must first familiarize with the baseline features (mutational status, transcripts, proteins/protein modifications, metabolites) and interactions between these components (gene–RNA, RNA–protein, protein–protein) across various cellular states [20][23]. Ex vivo drug sensitivity towards a panel of therapies can then be measured by monitoring the direction and strength of evolution of these interactive signaling events in all (non-) malignant cells. Miniaturizing the assay, for instance with chip technology would enable testing of multiple treatment conditions, while still providing sufficient multiparameter resolution on phenotypic and functional changes. These results could then be used by a medical board to integrate the functional finding (e.g., a ranked list of therapeutic options from most to least active in the tumor cells of the investigated patient) with baseline features and clinical parameters, such as tumor size, tumor location, extension of tumor resection and drug tolerability. Once all data is integrated, the most suitable drug/combination could be selected for each, individual patient. As anticipated, this procedure can be repeated once the GBM tumor recurs (Figure 1).
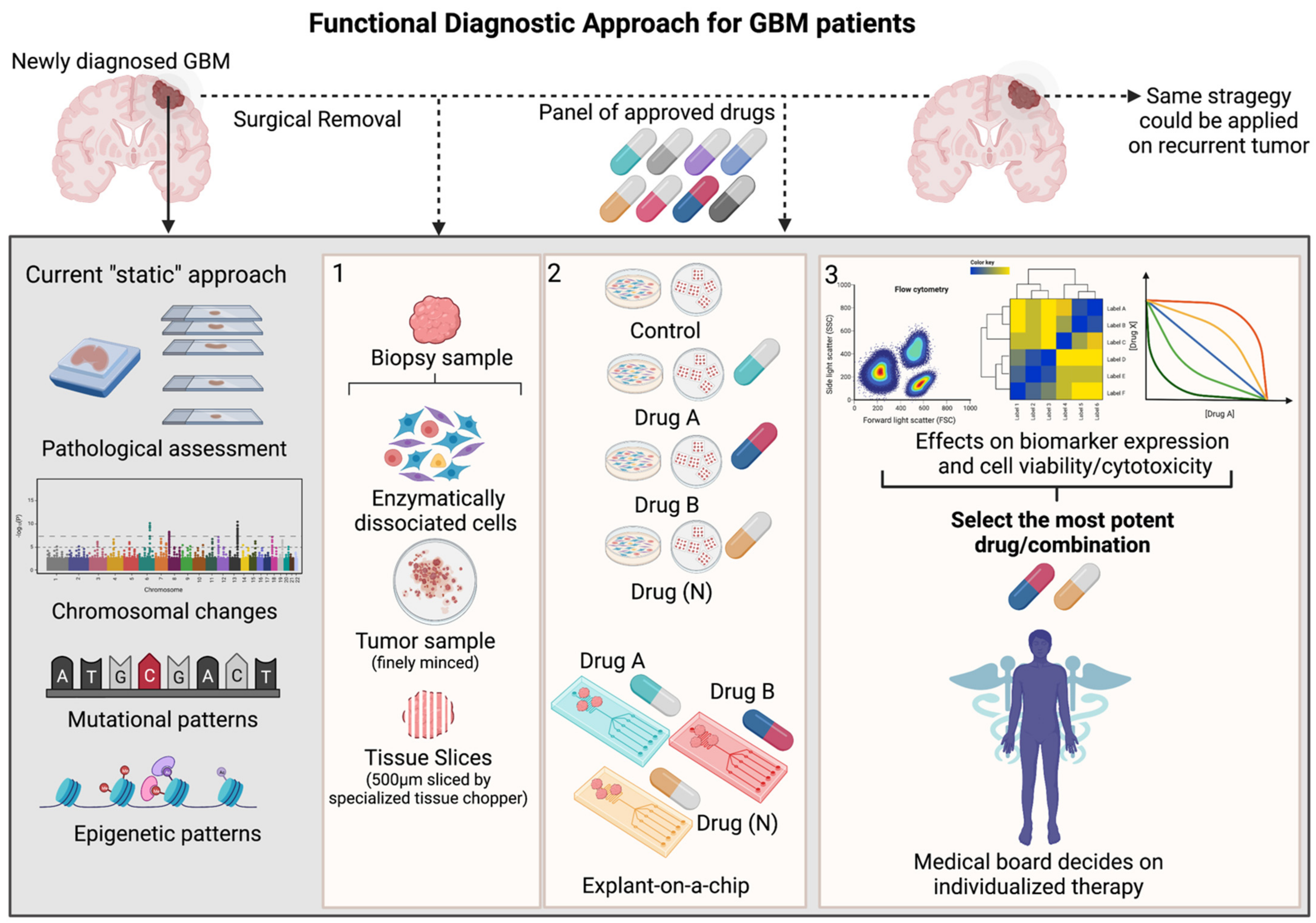
Figure 1. Schematic overview of functional diagnostic approach in GBM (Created with BioRender.com). During craniotomy, biopsy samples are routinely collected from newly diagnosed or recurrent GBM patients and pathologically assessed using standard clinical procedures, including immunohistochemical staining (IHC) of a handful of markers aiding histological grading, next generation sequencing and molecular analysis uncovering mutational patterns and epigenetic sequencing that measures the MGMT-promotor methylation status Although highly relevant, all these techniques offer only a single glance at the tumor’s baseline features (“static” measurement) and do not completely capture the intra-tumoral heterogeneity and therapeutic vulnerability of the patient’s tumor. To resolve this task, functional diagnostic is a personalized medicine strategy that makes use of live tumor samples derived from each individual patient. Panel 1: These biopsy samples can be enzymatically dissociated, minced or cut into fine tissue layers/slices. Panel 2: As such, these probes can be ex vivo treated with a panel of approved GBM-targeting therapies in cell culture flasks/plates or microfluidic chips. Panel 3: Various methods could be applied in order to optimally capture the effects of the given therapy on functional cellular features (cyto-toxic/-static events, various cellular states or cellular signaling pathways) relevant and corresponding to the given treatment. The output of these functional measurements would be a ranked list of most potent therapies, whereby a medical board could integrate this information together with histological, molecular measurements and clinical parameters. Finally, clinicians could decide on which therapy would be the most beneficial for each patient. This strategy could be applied on patients diagnosed with a recurrent tumor.
3. Tools and Methods for Functional Diagnostics
The development of functional diagnostic assays strongly depends on the availability of representative cancer models that maximally capture the genetic and phenotypic features of patients’ tumor. So far, in vitro cancer research has been relying on so-called conventional cancer cell lines [24], which, although easy to use and representative in broader disease terms, have important limitations including: (i) lack of predictive value with regard to activity in clinical trials; and (ii) display of major and irreversible alterations in biological properties, such as gains and losses of genetic information, alterations in growth and invasive properties and loss of biomarker expression compared to the original tumor [24]. The growing body of evidence of heterogeneity, along with technological advances and platforms for drug development, steered pre-clinical research towards models derived from diseased individuals, such as patient-derived cell lines (PDCLs), patient-derived organoids (PDOs) and patient-derived xenografts (PDX). For GBM, an armamentarium of such models has been developed, although the installation of optimal readouts to assess drug activity in either of them still remains challenging (Figure 2).
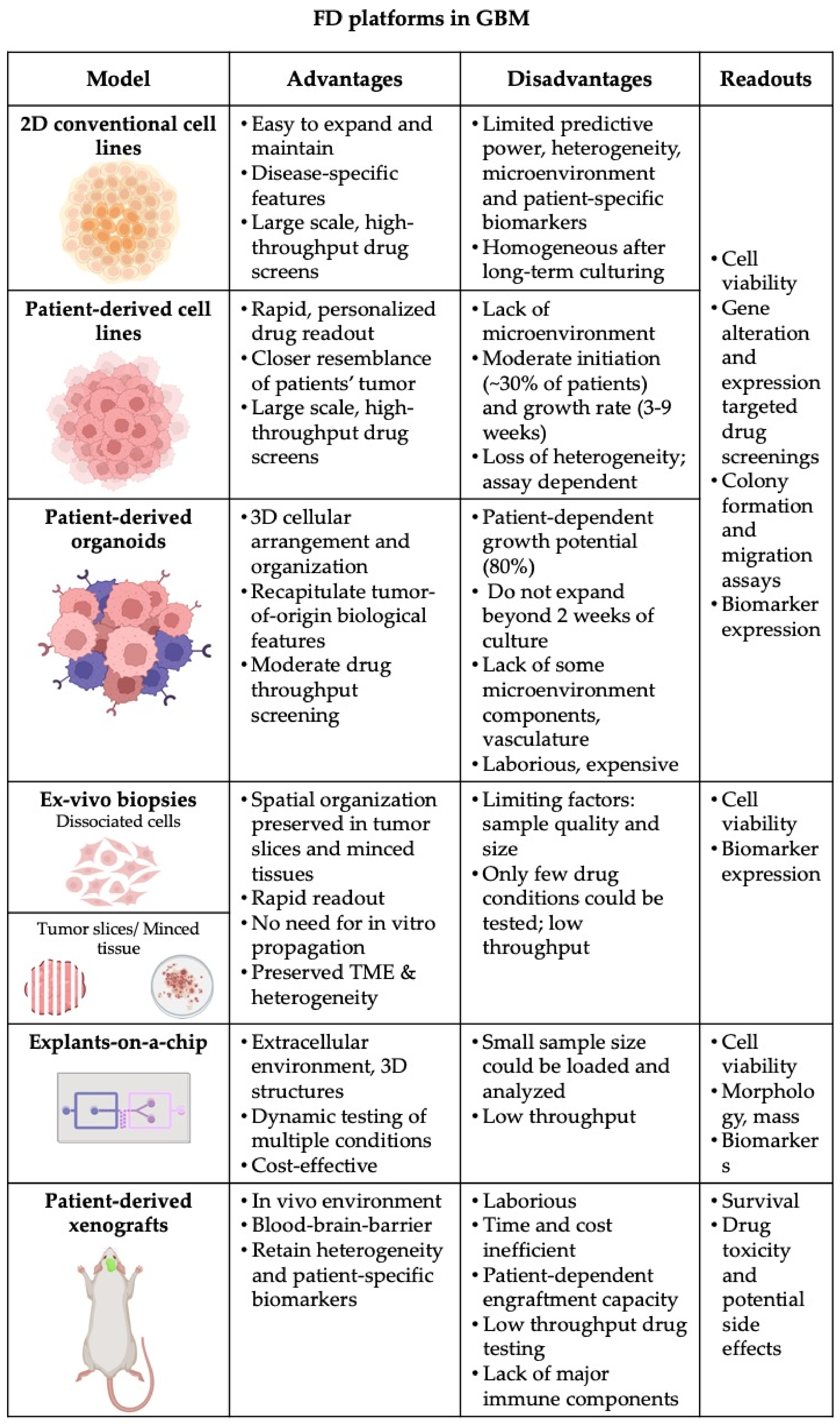
Figure 2. Summary of pre-clinical models and platforms, which could be used for functional testing in GBM. Advantages and disadvantages of each model together with potential assay readout are outlined (Created with Biorender.com).
Drug screening in PDX clinical trials were executed for various cancer types and solid tumors [25]. With this concept. it was confirmed that PDX models have the ability to predict trial responses, by evaluating predictive response biomarkers, map resistance mechanisms [25] and guide treatment decision making [24][26][27][28]. Patient-derived xenograft models for GBM are generated by direct transplantation of dissociated patient tumor material or tumor pieces. While a tendency for CNV-loss in heterotopic models has been suggested, orthotopic PDX models typically retain a close resemblance to the primary tumor [29]. Interestingly, studies confirm that the tumor-of-origin resemblance is highly dependent on the region from which the biopsy has been harvested, meaning that two PDX models generated from distinct regions of a single tumor could generate PDX models with dissimilar tumor subpopulations [29]. XENOGBM is a study currently evaluating the molecular analogy between the primary tumors of GBM patients and their corresponding PDX models (NCT02904525). PDX platforms are more advantageous over in vitro cultures as they retain 3D structural organization, clinical features, such as tumor invasiveness, vascularization, pseudopalisading necrosis and therapy-induced tumor evolution, and molecular features of the primary tumor, for instance crucial biomarkers such as EGFR expression, which is regularly suppressed by culturing conditions [30][31]. Furthermore, orthotopic PDX models provide the in vivo CNS environment enclosed behind the blood–brain barrier (BBB) allowing the direct evaluation of the penetration capacity and metabolomics of pharmacologic agents. Although seemingly superior over other models, PDXs still have several disadvantages for precision medicine in GBM. These models are laborious, time consuming and expensive in comparison to cell lines and organoids. The tumor take rate has been shown to be quite variable, meaning that PDX models would not be generated for all patients, or the number of models would be too limited in order to evaluate sufficient numbers of drug or drug combinations. Furthermore, the time between tumor engraftment and therapy decision may be too long for GBM patients. Finally, the use of immunodeficient mice largely hinders the interrogation of the role of the immune system in treatment responsiveness and general tumor biology.
In general, tumors including GBM, release cells and cellular content into the bloodstream or cerebrospinal fluid (CSF). These biomarkers are shed from the tumor residing site in form of circulating tumor cells (CTCs), proteins, cell-free nucleic acids and extracellular vesicles (EVs), accordingly systemized as liquid biopsies [32]. As such, liquid biopsies set ground for a rapid, noninvasive way for cancer diagnosis and prognostic markers [32]. Currently, liquid biopsies have gained clinical application for metastatic breast cancer [33], small cell lung cancer [34], prostate [35] and colorectal cancer [36] in the context of tumor diagnosis and longitudinal monitoring of therapy responses in both primary and metastasized tumors. Specifically, it has been shown that CTC count in peripheral blood correlates to therapy response. Advanced molecular profiling of these cells shows a high degree of concordance between genomic and transcriptomic profiles with the tumor of origin, making CTCs an excellent tool that could support personalized medicine approaches [37]. CTC-derived cell lines for various cancer types enabled CTC characterization and in vitro drug treatments, which may inform on the treatment susceptibility of the primary tumor and identify ways to inhibit metastasis [38]. This has been further corroborated by short-term ex vivo propagation of small cell lung cancer (SCLC) CTCs in culture from 23 patients. The CTC-derived cultures were in vitro treated with cisplatin and etopside and the results were correlated with individual responses from the respective patients. The results of this investigation showed correlation between response profiles of ex vivo expanded CTCs and three patients. Furthermore, this study highlights the ability of in vitro treated CTCs to accurately inform on innate and acquired chemo-resistance, based on patients’ treatment history and clinical outcomes [34]. A similar report emphasized the predictive accuracy of in vitro-treated CTCs and two respective patients diagnosed with head and neck cancer and treated with cisplatin [39].
Owing to their location, brain tumors are challenging for surgical resection. Even when the tumor is accessible, the invasive surgery and biopsy collection present a risk of swelling and neurological damage. As patients receive an MRI scan within 12 weeks of treatment, contrast-enhancing lesions that are revealed on the images can indicate tumor progression, but might also be caused by post-radiotherapy edema, termed as pseudoprogression, which can spontaneously resolve [40][41]. At the moment, there are no methods that could reliably differentiate between glioma progression and pseudoprogression, or longitudinally monitor disease and treatment effects. The validation of biomarkers from liquid biopsies that could aid GBM prognosis is steadily progressing [41]. Liquid biopsies can be collected from cerebrospinal fluid (CSF), as it is in close contact with the CNS and accumulates tumor-specific markers, but CSF collection through lumbar punction is an invasive procedure [40][41]. In this light, the minimally invasive procedure to obtain liquid biopsies from GBM patients is through blood draw, but one must assume that the BBB is compromised at the tumor site. BBB disruption and permeability increases, as GBM tumors invade and progress into the surrounding tissue [41]. Therefore, CTC enumeration or EVs detection can potentially complement current strategies for more precise prediction of GBM progression. At present, methods for optimal CTC isolation and detection are advancing for GBM [32]. Unlike other epithelial-derived cancers where strong surface expression of EpCAM is detected (such as breast, prostate cancer, pancreatic, colorectal and hepatocellular), RNA sequencing of GBM-derived CTCs revealed Wnt-activated stemness and enrichment of mesenchymal features [41][42]. Alternative methods for CTCs detection in GBM include: GFAP labelling, telomerase-based assay, FISH detection of aneuploidy of chromosome 8, CTC-iChip microfluidic platform, recombinant VAR2CSA Malaria Protein and hTERT-specific oHSV1 expressing GFP [41]. All these studies point out the clinical utility of CTCs and liquid biopsies in real-time disease monitoring, prediction of progression and even functional measurements [43]. However, the number of CTCs is genuinely low (1–10 cells per 10 mL blood; 1 cell per 109 blood cells); therefore, efficient CTC isolation which recapitulates intra-tumoral heterogeneity and enables functional assessment is still far beyond the reach of GBM patients [32][40][41].
Hence, the ideal model for rapid functional assessment of drug sensitivity in GBM would be a system which maximally preservers the native cellular integrity [44] and interaction of the tumor cells with the microenvironment [30]. This includes ex-vivo drug treatment of tumor slices [22] or cellular suspensions of freshly dissociated patients’ biopsies within hours post-surgery. Regarding GBM’s extensive heterogeneity and invasiveness, one must consider sampling from distinct tumor regions in order to gain an “as close as possible” perspective of the therapeutic vulnerabilities of the invading cells that are remaining after tumor debulking. In a recent proof-of-concept study of a single GBM patient, tumor material was harvested and analyzed with single-cell RNA sequencing and scATAC-seq. The leftover patient material was orthotopically transplanted into mice, which were then treated with standard-of-care therapy (irradiation and temozolomide). Subsequently, the patient tumor was harvested and analyzed at recurrence. This framework provided mechanistic genetic and epigenetic insights into therapy-driven evolution and identified potential druggable targets, therefore providing an approach for designing therapeutic regimens for GBM [45]. Yet another proof-of-principle study demonstrated the efficacy of drug screening human breast cancer cell lines through imaging mass cytometry, assessing more than 40 markers [46]. All these methodologies are facing technological challenges, which need to be improved, upscaled and validated in order to meet the needs of routine clinical practice.
An auspicious high-throughput drug screening methodology has emerged with microfluidic devices [47]. The chip technology closely mimics the extracellular environment, which in turn is capable of generating 3D structures of cells. Such a device was designed to recapitulate the complex vasculature of the BBB and track the transport of nanoparticles to GBM spheroids. Analogous permeability measurements were performed on orthotopic xenografts through intravital imaging, which matched the in vitro model [48]. Microfluidic devices are automated and multiplexed platforms where the controlled environment offers a way to monitor drug effects, such as cell viability, changes in cellular mass accumulation rate upon treatment and morphology [49] at multiple timepoints [47][50].
References
- Yabo, Y.A.; Simone, P. Cancer Cell Heterogeneity and Plasticity: A Paradigm Shift in Glioblastoma. Neuro-Oncology 2021, 24, 669–682.
- Zanders, E.D.; Svensson, F.; Bailey, D.S. Therapy for Glioblastoma: Is It Working? Drug Discov. Today 2019, 24, 1193–1201.
- Stupp, R.; Mason, W.; van den Bent, M.; Weller, M.; Fischer, B.; Taphoorn, M.; Belanger, K.; Brandes, A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. J. Neurooncol. 2005, 352, 987–989.
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the Extent of Resection with Survival in Glioblastoma. JAMA Oncol. 2016, 2, 1460.
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477.
- Ceccarelli, M.; Barthel, F.P.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Pagnotta, S.M.; Anjum, S.; Wang, J.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563.
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110.
- Kim, G.; Ko, Y.T. Small Molecule Tyrosine Kinase Inhibitors in Glioblastoma. Arch. Pharm. Res. 2020, 43, 385–394.
- Di Stefano, A.L.; Fucci, A.; Frattini, V.; Labussiere, M.; Mokhtari, K.; Zoppoli, P.; Marie, Y.; Bruno, A.; Boisselier, B.; Giry, M.; et al. Detection, Characterization, and Inhibition of FGFR-TACC Fusions in IDH Wild-Type Glioma. Clin. Cancer Res. 2015, 21, 3307–3317.
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 Inhibitors and Cancer Therapy. Radiother. Oncol. 2018, 126, 450–464.
- Carrassa, L.; Damia, G. DNA Damage Response Inhibitors: Mechanisms and Potential Applications in Cancer Therapy. Cancer Treat. Rev. 2017, 60, 139–151.
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma Targeted Therapy: Updated Approaches from Recent Biological Insights. Ann. Oncol. 2017, 28, 1457–1472.
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Shawn, M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401.
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410.
- Chen, X.; Wen, Q.; Stucky, A.; Zeng, Y.; Gao, S.; Loudon, W.G.; Ho, H.W.; Kabeer, M.H.; Li, S.C.; Zhang, X.; et al. Relapse Pathway of Glioblastoma Revealed by Single-Cell Molecular Analysis. Carcinogenesis 2018, 16, 872–876.
- Wang, L.; Babikir, H.; Müller, S.; Yagnik, G.; Shamardani, K.; Catalan, F.; Kohanbash, G.; Alvarado, B.; Di Lullo, E.; Kriegstein, A.; et al. The Phenotypes of Proliferating Glioblastoma Cells Reside on a Single Axis of Variation. Cancer Discov. 2019, 9, 1708–1719.
- Daniel, P.; Sabri, S.; Chaddad, A.; Meehan, B.; Jean-Claude, B.; Rak, J.; Abdulkarim, B.S. Temozolomide Induced Hypermutation in Glioma: Evolutionary Mechanisms and Therapeutic Opportunities. Front. Oncol. 2019, 9, 1–7.
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963.
- Shin, S.H.; Bode, A.M.; Dong, Z. Precision Medicine: The Foundation of Future Cancer Therapeutics. npj Precis. Oncol. 2017, 1, 12.
- Friedman, A.A.; Letai, A.; Fisher, D.E.; Flaherty, K.T. Precision Medicine for Cancer with Next-Generation Functional Diagnostics. Nat. Rev. Cancer 2015, 15, 747–756.
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.I.; Iwama, T.; Park, D.M. The Evidence of Glioblastoma Heterogeneity. Sci. Rep. 2015, 5, 7979.
- Zhao, W.; Dovas, A.; Spinazzi, E.F.; Levitin, H.M.; Banu, M.A.; Upadhyayula, P.; Sudhakar, T.; Marie, T.; Otten, M.L.; Sisti, M.B.; et al. Deconvolution of Cell Type-Specific Drug Responses in Human Tumor Tissue with Single-Cell RNA-Seq. Genome Med. 2021, 13, 1–15.
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21.
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in Vitro and in Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477.
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-Throughput Screening Using Patient-Derived Tumor Xenografts to Predict Clinical Trial Drug Response. Nat. Med. 2015, 21, 1318–1325.
- Vargas, R.; Gopal, P.; Kuzmishin, G.B.; DeBernardo, R.; Koyfman, S.A.; Jha, B.K.; Mian, O.Y.; Scott, J.; Adams, D.J.; Peacock, C.D.; et al. Case Study: Patient-Derived Clear Cell Adenocarcinoma Xenograft Model Longitudinally Predicts Treatment Response. NPJ Precis. Oncol. 2018, 2, 14.
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-Derived Xenografts Effectively Capture Responses to Oncology Therapy in a Heterogeneous Cohort of Patients with Solid Tumors. Ann. Oncol. 2017, 28, 2595–2605.
- Inoue, A.; Deem, A.K.; Kopetz, S.; Heffernan, T.P.; Draetta, G.F.; Carugo, A. Current and Future Horizons of Patient-Derived Xenograft Models in Colorectal Cancer Translational Research. Cancers 2019, 11, 1321.
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse Models of Glioblastoma for the Evaluation of Novel Therapeutic Strategies. Neuro-Oncol. Adv. 2021, 3, 1–16.
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1–16.
- Schulte, A.; Günther, H.S.; Martens, T.; Zapf, S.; Riethdorf, S.; Wülfing, C.; Stoupiec, M.; Westphal, M.; Lamszus, K. Glioblastoma Stem-like Cell Lines with Either Maintenance or Loss of High-Level EGFR Amplification, Generated via Modulation of Ligand Concentration. Clin. Cancer Res. 2012, 18, 1901–1913.
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A.A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878.
- Ivanova, E.; Ward, A.; Wiegmans, A.P.; Richard, D.J. Circulating Tumor Cells in Metastatic Breast Cancer: From Genome Instability to Metastasis. Front. Mol. Biosci. 2020, 7, 1–11.
- Lee, H.L.; Chiou, J.F.; Wang, P.Y.; Lu, L.S.; Shen, C.N.; Hsu, H.L.; Burnouf, T.; Ting, L.L.; Chou, P.C.; Chung, C.L.; et al. Ex Vivo Expansion and Drug Sensitivity Profiling of Circulating Tumor Cells from Patients with Small Cell Lung Cancer. Cancers 2020, 12, 3394.
- Ried, K.; Tamanna, T.; Matthews, S.; Eng, P.; Sali, A. New Screening Test Improves Detection of Prostate Cancer Using Circulating Tumor Cells and Prostate-Specific Markers. Front. Oncol. 2020, 10, 582.
- Jiang, M.; Jin, S.; Han, J.; Li, T.; Shi, J.; Zhong, Q.; Li, W.; Tang, W.; Huang, Q.; Zong, H. Detection and Clinical Significance of Circulating Tumor Cells in Colorectal Cancer. Biomark. Res. 2021, 9, 85.
- Lallo, A.; Schenk, M.W.; Frese, K.K.; Blackhall, F.; Dive, C. Circulating Tumor Cells and CDX Models as a Tool for Preclinical Drug Development. Transl. Lung Cancer Res. 2017, 6, 397–408.
- Smit, D.J.; Pantel, K.; Jücker, M. Circulating Tumor Cells as a Promising Target for Individualized Drug Susceptibility Tests in Cancer Therapy. Biochem. Pharmacol. 2021, 188, 114589.
- Lin, K.C.; Ting, L.L.; Chang, C.L.; Lu, L.S.; Lee, H.L.; Hsu, F.C.; Chiou, J.F.; Wang, P.Y.; Burnouf, T.; Ho, D.C.Y.; et al. Ex Vivo Expanded Circulating Tumor Cells for Clinical Anti-Cancer Drug Prediction in Patients with Head and Neck Cancer. Cancers 2021, 13, 6076.
- Müller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating Biomarkers in Patients with Glioblastoma. Br. J. Cancer 2020, 122, 295–305.
- Zhang, H.; Yuan, F.; Qi, Y.; Liu, B.; Chen, Q. Circulating Tumor Cells for Glioma. Front. Oncol. 2021, 11, 1–9.
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating Tumor Cells: Biology and Clinical Significance. Signal Transduct. Target. Ther. 2021, 6, 404.
- Crook, T.; Gaya, A.; Page, R.; Limaye, S.; Ranade, A.; Bhatt, A.; Patil, S.; Kumar, P.; Patil, D.; Akolkar, D. Clinical Utility of Circulating Tumor-Associated Cells to Predict and Monitor Chemo-Response in Solid Tumors. Cancer Chemother. Pharmacol. 2021, 87, 197–205.
- Rosenberg, S.; Verreault, M.; Schmitt, C.; Guegan, J.; Guehennec, J.; Levasseur, C.; Marie, Y.; Bielle, F.; Mokhtari, K.; Hoang-Xuan, K.; et al. Multi-Omics Analysis of Primary Glioblastoma Cell Lines Shows Recapitulation of Pivotal Molecular Features of Parental Tumors. Neuro. Oncol. 2017, 19, 219–228.
- Park, J.H.; Feroze, A.H.; Emerson, S.N.; Mihalas, A.B.; Keene, C.D.; Cimino, P.J.; de Lomana, A.L.G.; Kannan, K.; Wu, W.-J.; Turkarslan, S.; et al. A single-cell based precision medicine approach using glioblastoma patient-specific models. bioRxiv 2021, 1–28.
- Bouzekri, A.; Esch, A.; Ornatsky, O. Multidimensional Profiling of Drug-Treated Cells by Imaging Mass Cytometry. FEBS Open Bio 2019, 9, 1652–1669.
- Sun, J.; Warden, A.R.; Ding, X. Recent Advances in Microfluidics for Drug Screening. Biomicrofluidics 2019, 13, 1–13.
- Straehla, J.P.; Hajal, C.; Safford, H.C.; Offeddu, G.S. A Predictive Micro Fl Uidic Model of Human Glioblastoma to Assess Traf Fi Cking of Blood—Brain Barrier-Penetrant Nanoparticles. Proc. Natl. Acad. Sci. USA 2022, 119, e2118697119.
- Stevens, M.M.; Maire, C.L.; Chou, N.; Murakami, M.A.; Knoff, D.S.; Kikuchi, Y.; Kimmerling, R.J.; Liu, H.; Haidar, S.; Calistri, N.L.; et al. Drug Sensitivity of Single Cancer Cells Is Predicted by Changes in Mass Accumulation Rate. Nat. Biotechnol. 2016, 34, 1161–1167.
- Stockslager, M.A.; Malinowski, S.; Touat, M.; Yoon, J.C.; Geduldig, J.; Mirza, M.; Kim, A.S.; Wen, P.Y.; Chow, K.H.; Ligon, K.L.; et al. Functional Drug Susceptibility Testing Using Single-Cell Mass Predicts Treatment Outcome in Patient-Derived Cancer Neurosphere Models. Cell Rep. 2021, 37, 109788.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
23 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No