Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Oluwakemi Ebenezer | -- | 5371 | 2022-08-15 23:30:22 | | | |
| 2 | Camila Xu | Meta information modification | 5371 | 2022-08-16 03:27:35 | | | | |
| 3 | Camila Xu | Meta information modification | 5371 | 2022-08-16 04:47:04 | | | | |
| 4 | Camila Xu | Meta information modification | 5371 | 2022-08-16 08:13:43 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ebenezer, O.; Jordaan, M.A.; Carena, G.; Bono, T.; Shapi, M.; Tuszynski, J.A. Five-Membered Heterocyclic Compounds. Encyclopedia. Available online: https://encyclopedia.pub/entry/26164 (accessed on 08 August 2026).
Ebenezer O, Jordaan MA, Carena G, Bono T, Shapi M, Tuszynski JA. Five-Membered Heterocyclic Compounds. Encyclopedia. Available at: https://encyclopedia.pub/entry/26164. Accessed August 08, 2026.
Ebenezer, Oluwakemi, Maryam Amra. Jordaan, Gea Carena, Tommaso Bono, Michael Shapi, Jack A. Tuszynski. "Five-Membered Heterocyclic Compounds" Encyclopedia, https://encyclopedia.pub/entry/26164 (accessed August 08, 2026).
Ebenezer, O., Jordaan, M.A., Carena, G., Bono, T., Shapi, M., & Tuszynski, J.A. (2022, August 15). Five-Membered Heterocyclic Compounds. In Encyclopedia. https://encyclopedia.pub/entry/26164
Ebenezer, Oluwakemi, et al. "Five-Membered Heterocyclic Compounds." Encyclopedia. Web. 15 August, 2022.
Copy Citation
Heterocyclic compounds are a class of compounds of natural origin with favorable properties and hence have major pharmaceutical significance. They have an exceptional adroitness favoring their use as diverse smart biomimetics, in addition to possessing an active pharmacophore in a complex structure. This has made them an indispensable motif in the drug discovery field. Heterocyclic compounds are usually classified according to the ring size, type, and the number of heteroatoms present in the ring.
heterocyclic compounds
nitrogen containing compounds
biological evaluations
1. Introduction
Heterocyclic compounds are compounds with a cyclic ring bearing carbon and other elements, such as oxygen, nitrogen, and sulfur. The simplest of the five-membered heterocyclic compounds are pyrrole, furan, and thiophene, and the compounds contain a single heteroatom. Notably, heterocyclic compounds are the preferred class of compounds of natural origin with pharmaceutical significance. They have an exceptional adroitness to be used as diverse smart biomimetics and an active core in a complex structure, which has made them an indispensable motif in the pharmaceutical field. Several heterocyclic derivatized compounds are known. These numbers continue to expand rapidly, as heterocyclic substitutions can alter the degree of ionization of compounds in the physiological pH, resulting in changes in their basicity and lipophilicity, leading to substantial differences in pharmacokinetic properties [1][2].
2. Five-Membered Heterocyclic Compounds
2.1. Triazole
Heterocyclic systems continue to generate considerable interest due to their broad spectrum of biological activities. Triazole and its derivatives have increased in importance as they represent the structural characteristics of many bioactive compounds. They are known to be included in the structure of many medications, namely, itraconazole, fluconazole, voriconazole, ribavirin, mubritinib, and posaconazole, amongst others. The triazole ring features three nitrogen atoms in the five-membered aromatic ring and is significantly isomeric based on the placement of nitrogen atoms in the ring. Triazole and its derivatives can interact with various enzymes and receptors in the biological system through the diversity of non-covalent interactions, thereby presenting versatile biological activities [3]. Two significant forms of triazole include 1,2,3-triazole and 1,2,4-triazole. Extensive research on triazole and its derivatives has shown the major pharmacological importance of this heterocyclic nucleus.
2.2. 1,2,3-Triazole
Riu et al. [4] designed and synthesized new and improved benzotriazole–acrylonitrile derivatives incorporating two halogen atoms in positions 5′ and 6′ on the benzotriazole moiety. The compounds were further subjected to biological evaluation. Compound 1 was the most potent in the new series of derivatives. The in vitro XTT assay, flow cytometry analysis, and immunostaining performed on HeLa cancer cells treated with 1 displayed a significant antiproliferative effect, with an IC50 value of 3.2 µM. It was demonstrated to block the cells in G2/M-phase, and subsequently cause cell division defects. Additionally, β-tubulin staining validated the microtubule as being a potential molecular target of 1, while the colchicine competition assay indicated that compound 1 vies with colchicine for the binding site on tubulin. Kasemsuk et al. [5] synthesized a novel series of acanthoic acid analogues by substituting a carboxyl functional group of acanthoic acid with methyl ester hybrids bearing a triazole ring through esterification and the CuAAC reaction and considered their cytotoxic activity against cholangiocarcinoma cell lines. Among the evaluated compounds, 2 showed the most significant activity with an IC50 value of 18 μM against the KKU-213 cell line, which was eight-fold more effective than acanthoic acid. An assessment of anti-inflammatory, ulcerogenic, platelet activation activities, and the molecular docking studies of COX-2 and P-selectin of the 1,4-diaryl-1,2,3-triazole hybrids has been recently published [6]. The analogs 3–5 exhibited anti-inflammatory activity and lacked the induction of gastric lesions in mice when compared to the reference drug, indomethacin. The reduction of polymorphonuclear cells’ influx into the peritoneal cavity caused by carrageenan indicated that these active compounds could favor a dual inhibition of COX-2 and 5-LOX enzymes. The hybrids caused a reduction in the expression of P-selectin, which may be liable to mitigate inflammation and thromboembolic events. The molecular docking study with P-selectin showed crucial interactions with the amino acid residue Tyr 48 (see Figure 1).
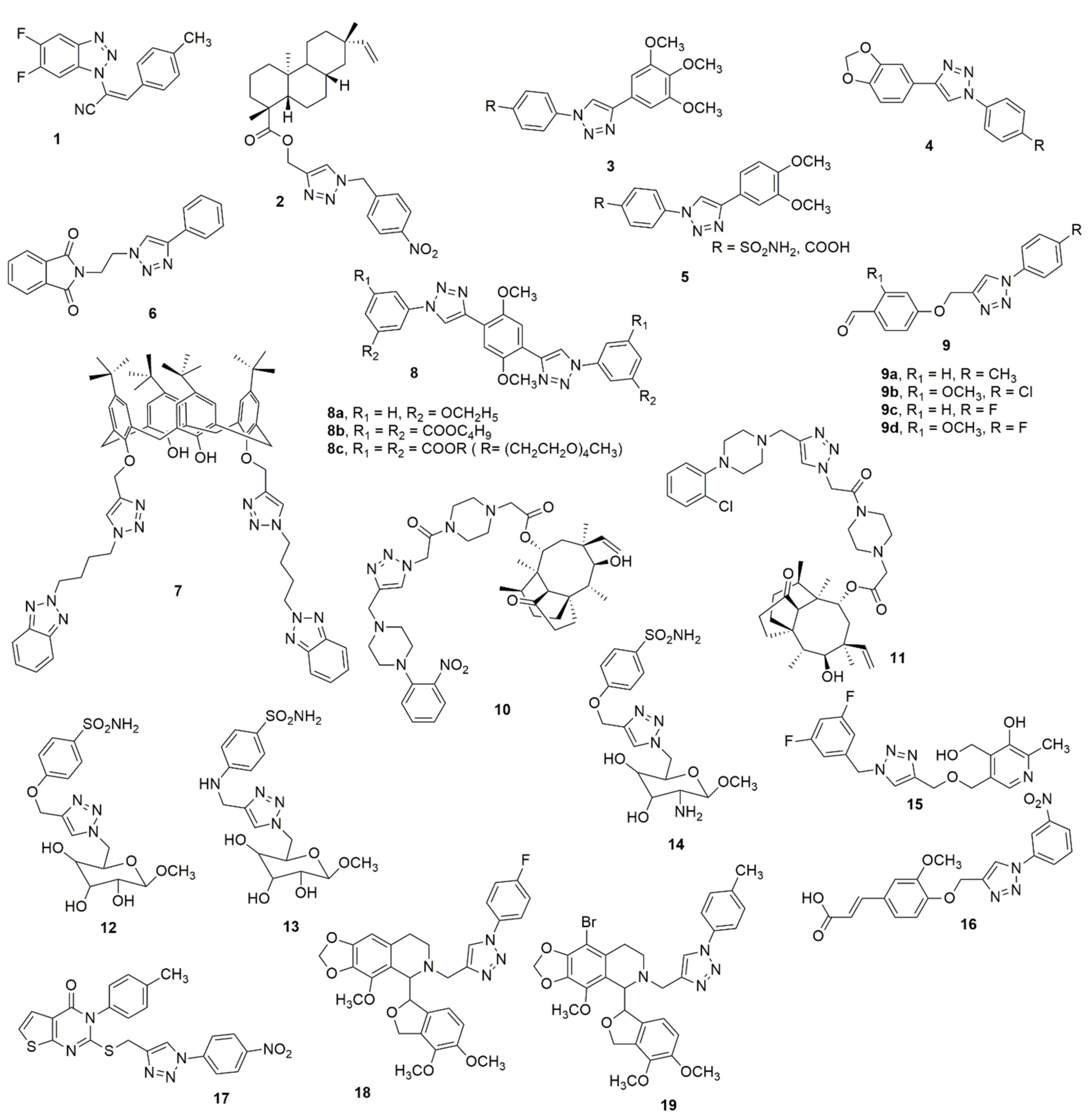
Figure 1. Structures of most active analogues of 1,2,3-triazole molecule.
Holanda et al. [7] utilized the click chemistry approach to efficiently synthesize alkyl-substituted phthalimide 1H-1,2,3-triazole derivatives and evaluated their leishmanicidal potential against Leishmania amazonensis and Leishmania braziliensis. Compound 6 selectively inhibited the increase and existence of promastigote and amastigote forms of L. amazonensis and L. braziliensis. The molecular docking study indicates that compound 6 is a potential inhibitor of parasite sterol 14α-demethylase due to its interaction with the heme groups of this enzyme. The amalgamation of benzotriazoles and calixarenes through click chemistry produces a new class of p-tert-butyl-calix [8] arene tethered benzotrizolyl dendrimers, which has been reported together with their biological evaluation [9]. Compound 7 was found to be the most potent antibacterial and anti-biofilm agent against drug-resistant and slime-generating organisms with no detected cytotoxicity to the mammalian cell line. Melah et al. [10] reported the design, synthesis, and in vitro antiproliferative activities of novel mono- and bis-1,2,3-triazole molecular hybrids. Compound 8a exhibited potent activity against HepG-2 when compared to the standard reference anticancer drug doxorubicin whereas compounds 8b and 8c demonstrated significant toxicity on the RPE-1 human normal cells. Sahin et al. [11] reported the design and synthesis of several 1,2,3-triazole compounds with aldehyde functional groups. The synthesized compounds were further examined for their antioxidant, anti-cancer, and α-amylase enzyme activity. The DPPH radical scavenging studies showed that all the compounds have a higher activity than the standard BHT and β-carotene. Compound 9a displayed almost the same scavenging effect with β-carotene and BHT. Compound 9b (IC50 = 165 μg/mL) was almost 5.5-fold superior to acarbose regarding its α-amylase inhibition activity. The synthesized compounds were screened for anti-cancer activities against the HeLa cell line. Compound 9c and 9d with IC50s of 50.12 and 57.07 μg/mL, respectively, displayed mild antitumor activity when compared to cisplatin against the HeLa cell line (see Figure 1).
A novel series of pleuromutilin derivatives bearing piperazine and 1,2,3-1H-triazole structures were synthesized by click chemistry methodology under mild conditions [12]. The compounds were further investigated for their MIC and MBC against methicillin-resistant S. aureus (ATCC 43300), S. aureus (ATCC 29213), S. aureus (AD3), S. aureus (144), and E. coli (ATCC 25922). Compounds 10 and 11 displayed more significant antibacterial activity than other compounds. Compound 10 exhibited rapid kinetics of its bactericidal activity against MRSA and had a longer PAE than tiamulin. The in vivo antibacterial effectiveness of 11 was further studied in a neutropenic murine thigh infection model. The outcomes revealed that compound 10 exhibited more effective in vivo antibacterial activity than tiamulin. In addition, 10 exhibited low to moderate repressing effects on CYP1A2, CYP2E1, CYP2D6, and CYP3A4 enzymes. The design, synthesis, and biological evaluation of a panel of novel aromatic sulfonamides linked to a hydrophilic sugar-tail moiety using rigid 1,2,3-triazole as a spacer have been reported [13]. The newly designed compounds were investigated in vitro and an efficient inhibition against all three CA isoforms, especially the tumor-associated hCA IX, was observed. All the glycoconjugate sulfonamide derivatives exhibited superior inhibitory activity. Compound 12 was the most effective and selective inhibitor of hCA IX with an inhibitory constant (IC50) value of 7 nM, being four-fold superior to acetazolamide (AAZ) whose IC50 value is 30 nM. In both hypoxic and normoxic conditions, almost all the compounds exhibited moderate antiproliferative activities against two cancer cell lines (HT-29 and MDA-MB-231). Notably, 12 exhibited superior antitumor activity and cytotoxic activity. In addition, the combined therapy evaluation found noticeable decreases (20–35%) in doxorubicin IC50 values in MDA-MB-231 cancer cells in a hypoxic environment in the presence of compounds 12–14, carbonic anhydrase inhibitors, when compared to single therapy (doxorubicin) (see Figure 1).
Multi-target natural product-pyridoxine-based derivatives were designed, synthesized, characterized, and evaluated as potential anti-Alzheimer agents [14]. Among the tested compounds, 15 acted as a potent acetylcholinesterase (AChE) inhibitor, (IC50 = 1.56 ± 0.02 mM) and exhibited antioxidant activity, having an ORAC-FL value of 1.21 ± 0.28, which is comparable to Trolox. The docking studies showed interactions between the peripheral anionic site of the enzyme (PAS site) with the hydrophobic amino acids Tyr 124 and Phe 338 and the triazole nucleus of 15. Compound 16 has been reported to selectively inhibit carbonic anhydrase IX (CAIX) with an IC50 value of 24 nM [15]. The in silico analysis revealed the binding of 16 with the catalytically significant amino acid residues of CAIX. Additionally, cell-based studies showed that 16 prevents the activity of CAIX, reduces the epithelial-to-mesenchymal transitions, induces apoptosis, and obstructs cell migration and colonization potential of cancer cells (see Figure 1).
Suryanarayana et al. [16] reported the synthesis of thieno[2,3-d]-pyrimidine fused 1,2,3-triazole scaffolds and their antioxidant activity. Compound 17 exhibited good antioxidant activity against DPPH scavenging with an IC50 value of 8.161 μM, as compared to the standard drug ascorbic acid (IC50 = 3.073 μM). Notably, electron-removal groups in the para-position of the derivatives provided excellent scavenging capacity for all three scavenging methods when compared to the electron-donating groups. Twenty novel 1,2,3-triazole noscapine derivatives have been synthesized using noscapine as a precursor and were further evaluated for their biological activity [17]. Interestingly the combination of computational and experimental evaluation revealed two potent compounds 18 and 19, (KD = 21.5 ± 6.15 and 36.9 ± 4.24 nM, respectively) compared to noscapine (KD = 579.0 ± 18.7 nM) (see Figure 1).
In an attempt to introduce new scaffolds as potent α-glucosidase inhibitors, Sepehri et al. [18] reported a new series of acridine-9-carboxamide-1,2,3-triazole-N-phenylacetamide derivatives. They were screened for their in vitro α-glucosidase inhibitory activities. Among the screened compounds, 20 exhibited a superior potency with an IC50 of 80.3 ± 0.9µM compared to the standard drug acarbose (IC50 = 750.0 ± 10.5µM). Cherif et al. [19] designed and synthesized some new hybrid compounds through the amalgamation of a pyranopyrimidinone moiety with 1,2,3-triazole pharmacophore via 1,3-dipolar cycloaddition using different arylazides. Compounds 21a–21d showed strong capacities of cholinesterase inhibition with IC50 values of 6.7 ± 0.2, 8.4 ± 0.4, 7.8 ± 0.2 and 9.1 ± 0.1 µM, respectively. Compound 22 has been reported to be a feasible positron emission tomography (PET) probe that can offer a better understanding of the ASGPR (asialoglycoprotein receptor)-related liver disease [20] (see Figure 2).
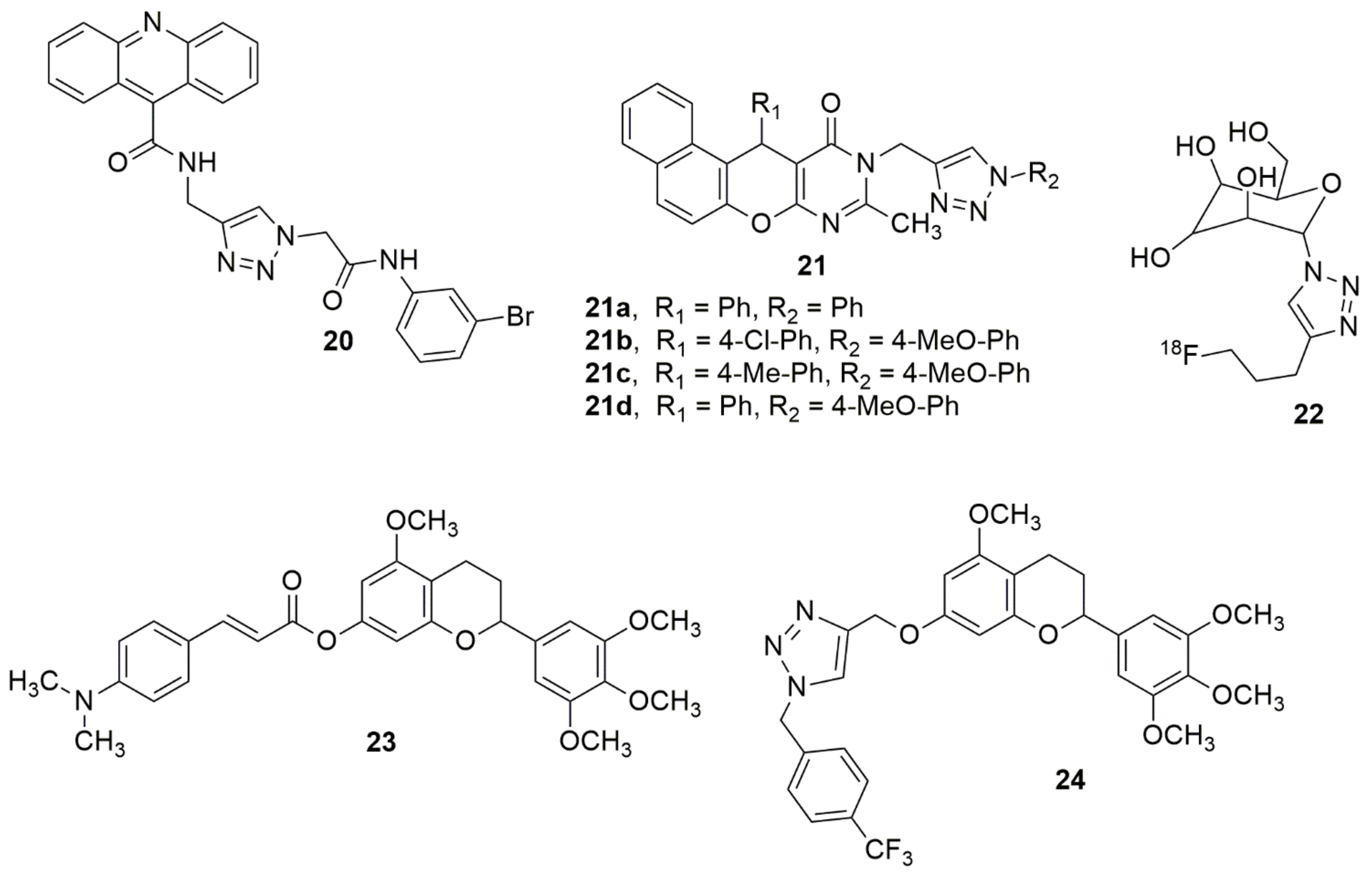
Figure 2. Chemical structures of 1,2,3-triazole hybrids with anti-diabetics (20) and anti-Alzheimer activities (21,23,24).
Shi and co-workers designed and synthesized 7-O-modified galloyltricetiflavan hybrids containing cinnamate, benzoate, phenyl sulfonate, and 1,2,3-triazole scaffolds [21]. Meanwhile, all synthesized compounds were examined for the inhibition of AChE/BuChE (butyrylcholinesterase) and anti-Aβ aggregation activity. Among the evaluated compounds, 23 exhibited the best inhibition of Aβ aggregation (78.81% at 20 μM), and superior AChE inhibitory potencies (IC50, 0.56 μM). Compound 24 exhibited the highest BuChE activity (IC50, 5.77 μM). Compounds 23 and 24 exhibited high potent protective capabilities than Trolox against H2O2- induced SH-SY5Y cell injuries. The observed potent compounds lacked visible toxicity in SH-SY5Y cells and could slightly increase SHSY5Y cell viabilities. Hence, 23 and 24 were reported as promising multi-functional agents for the treatment of Alzheimer’s disease (see Figure 2).
Tangadanchu et al. [22] designed, synthesized, and evaluated a series of eighteen new 1,2,3-triazole compounds and evaluated their sphingosine kinase-2 (SphK2) inhibitory activity using an ADP-Glo kinase assay. The in vivo anti-tumor bioactivity was further explored. Many of the screened compounds exhibited potent selectivity for SphK2 over SphK1. Compounds 25a, 26a–26c, 27a, and 27b were potent towards SphK2 with IC50 values of 0.234, 0.266, 0.254, 0.248, 0.261, and 0.269 µM, respectively, whereas the compounds 25a, 25d, 25e, 26b–26f, 27a–27c showed a high selectivity for SphK2 versus SphK1. In addition, compounds 25b–25c, and 27e exhibited superior antitumor activity for the human malignant glioblastoma tumor U-251 MG cell line when compared to ABC294640 (see Figure 3).
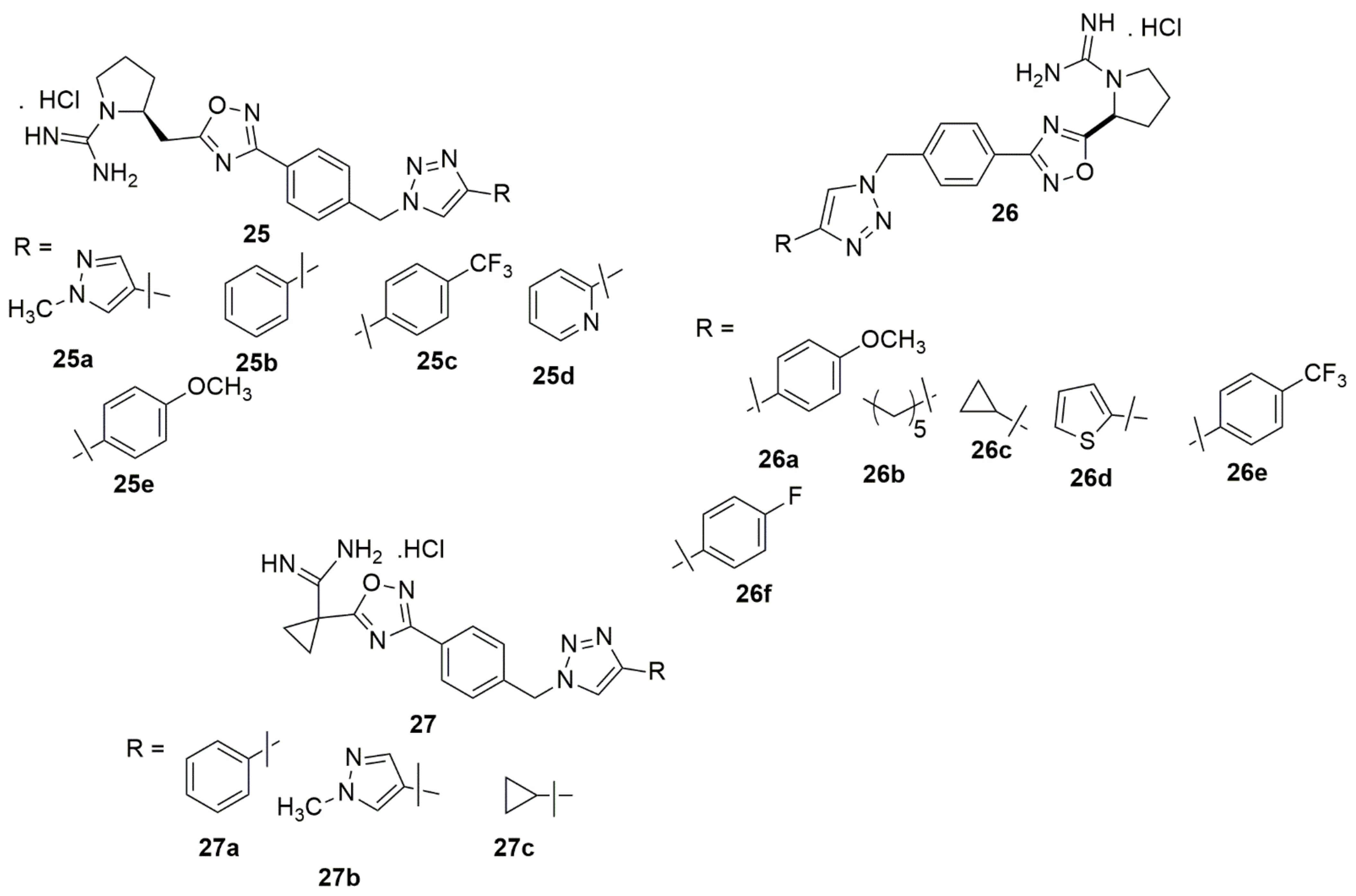
Figure 3. New 1,2,3-triazole series evaluated for SphK2 inhibitory activity using an ADP-Glo kinase assay.
Compound 28 has been reported to exhibit potent antiproliferative activity in U251 cells with an IC50 value of 0.94 µM, and it significantly inhibited the colony formation and migration of U251 cells [23]. Chaidam et al. [24] designed and synthesized a series of novel 1,6-bis-triazole-2,3,4-tri-O-benzyl-α-D-glucoside derivatives. The synthesized compounds were screened for their anti-diabetic activity. Among the examined compounds, 29 showed superior inhibitory activity with IC50 values of 3.73 μM, which was 39-fold higher than that of acarbose. Notably, the presence of the ester functional group and menthol moiety played a significant role in its biological activity due to increasing polarity, which enhanced binding against α-glucosidase. The library of the cationic tetrahydroisoquinoline–triazole compounds has been synthesized using the copper(II)-catalyzed azide–alkyne cycloaddition [25]. The compounds were evaluated for their antibacterial activity. Compound 30 potently inhibits Gram-positive pathogens and M. tuberculosis. The potent compound inhibited M. tuberculosis H37Rv at 6 μg/mL MIC. Compound 30 resulted in lysis and a bulging/swelling phenotype, suggesting compound 30 may target cell wall or membrane homeostasis. The cell passage test demonstrated that S.aureus did not develop resistance against 30 even at sub-inhibitory concentrations. A new series of quinazoline–triazole hybrid compounds have been designed, synthesized and evaluated for their anti-AChE activity by Le-Nhat-Thuy and co-workers [26]. Most of the synthesized compounds showed moderate to good AChEI activity. Among the evaluated compounds, N-benzyl-6-((1-(2-nitrophenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinazolin-4-amine 31 was shown to have the highest inhibitory activity with an IC50 value of 0.23 μM (see Figure 4).
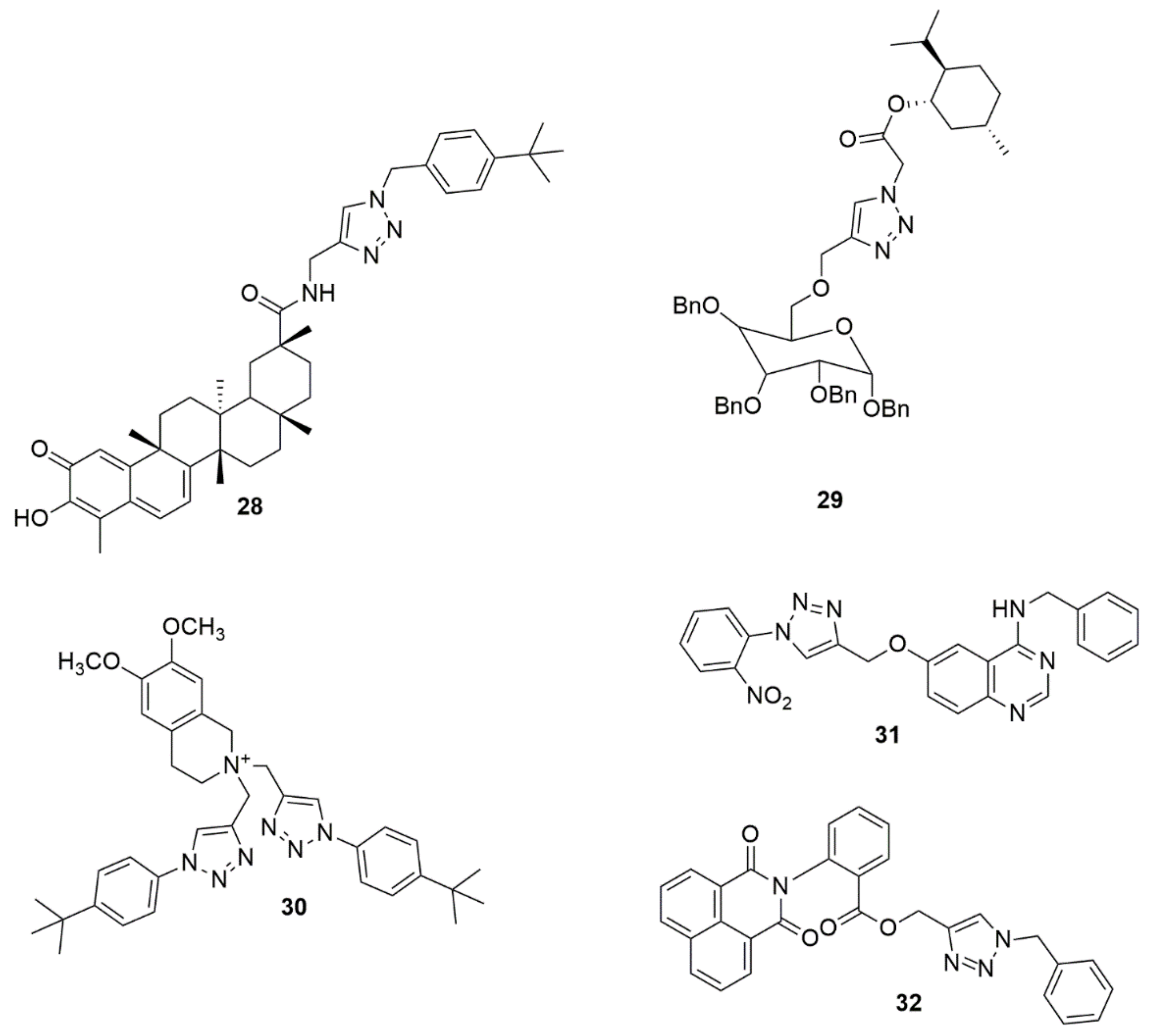
Figure 4. Chemical structures of 1,2,3-triazole hybrids with promising biological activities.
Bengam et al. [27] designed and synthesized novel naphthalimide-1,2,3-triazole tethered heterocycles and evaluated their in vitro anti-inflammatory properties. Among the compounds tested, 32 displayed inhibitions comparable to the reference compound (diclofenac sodium). The compound 32 showed 95.25% inhibition, and the reference drug showed 97.89% inhibition at 200 μM. The molecular docking analysis showed that the triazole ring 32 hydrogens bonded with the amine group of TYR385 and the amine group of TRP387 with the carbonyl group of anthranilic moiety (see Figure 4).
Hosseini et al. [28] used a molecular hybridization strategy to design a novel series of naphthoquinone derivatives bearing an acetamide–triazole moiety as novel AChE and BuChE inhibitors. Among the synthesized compounds evaluated for biological activity, 33 with an ortho-chlorine substituent exhibited the most potent AChE and BuChE activity with Ki values of 10.16 and 8.04 nM, respectively, compared to the standard compound Tacrine (Ki = 70.61 and 64.18 nM). Notably, the insertion of a chlorine atom at the ortho position enhanced the ChEs’ inhibition. Compound 33 was well fitted in the AChE and BuChE binding pocket via strong hydrogen bond interactions with the significant residue of each enzyme. Synthesis of a series of novel 1,2,3-triazole tethered chalcone derivatives and their cytotoxic activity against the human breast cancer cell line (MCF-7), cervical cancer (HeLa), and MDA-MB-231 cell lines have been reported [29]. In vitro cytotoxic activity evaluated using an MTT assay showed that all the synthesized compounds exhibited moderate to substantial cytotoxic activity. Compounds 34, 35, and 36 exhibited potent cytotoxic activity with IC50 values lower and comparable to cisplatin. Compounds 35 showed the best cytotoxic activity on MCF-7, with IC50 values of 1.27 and 0.02 µM at 24 and 48 h, respectively. Compounds with a chloro and methoxy substituent at different positions displayed promising activity. Abdel-Hafez and co-workers [30] designed, synthesized and hybridized acridine and coumarin derivatives, and evaluated their in vitro cancer cell growth inhibition activity. Among the evaluated compounds, 37 presented a good anticancer profile against MCF7 and DU-145 with IC50, values of 2.7 and 26.1 µM, respectively, comparable to doxorubicin (IC50 values = 2.0 and 14.2 µM). Compound 37 displayed greater inhibitory activity against topoisomerase (IIB) (IC50, 0.52 µM) when compared with doxorubicin (IC50 = 0.83 µM). The novel compounds 38a and 38b have been reported as DPP-4 inhibitors with IC50 values of 28 and 14 nM, respectively [31] (see Figure 5).
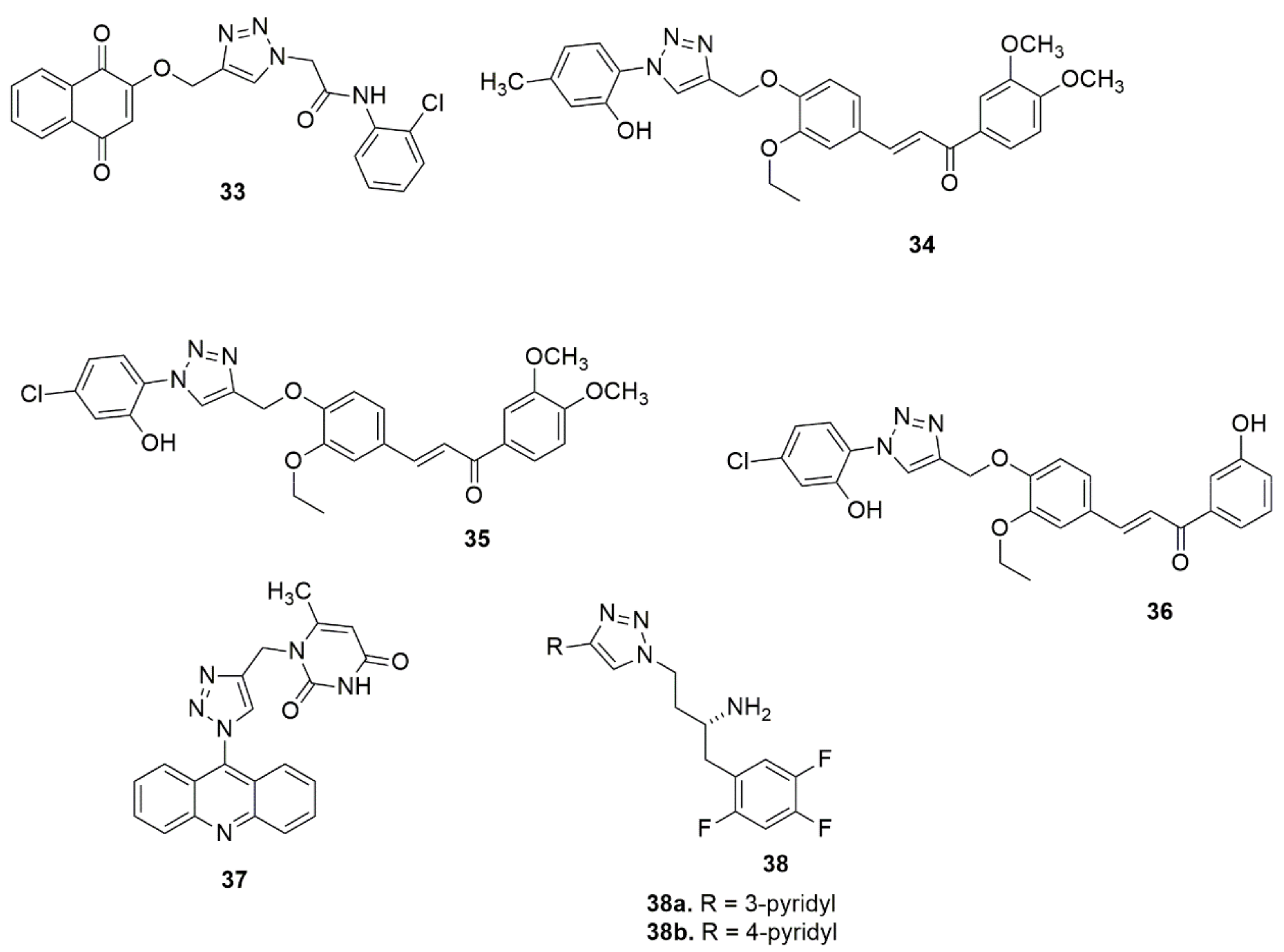
Figure 5. Chemical structures of 1,2,3-triazole hybrids with biological activities.
2.3. 1,2,4-Triazole
A novel series of 3-aryl-6-(N-methylpiperazin)-1,2,4-triazolo [3,4-a]phthalazines have been synthesized through a facile and economical one-pot copper-catalyzed method from 4-chloro-1-phthalazinyl-arylhydrazones as potential anticancer agents [32]. Most of the evaluated compounds showed anticancer activity against PC-3, MCF-7, and SKBr3 cancer cell lines. Interestingly, 39 displayed apparent anticancer activity with reduced toxicities, and appropriate selectivity indexes, and acted as potassium channel blockers. Meanwhile, the fused triazolo-phthalazine hybrid and the NO2 substituent enhanced the biological activity. Compound 40 was reported to be the most effective inhibitor of CB1 activity (0.644 µM) and showed the most effective selectivity of CB2/CB1 (>311) [33]. However, the lack of penetration of 40 through the blood–brain barrier similar to Rimonabant in the MDCK-mdr1 permeability analysis can result in a secondary effect on the CNS. This is apparently caused by the small, obstructed, hydrophobic cyclopropyl group of 1,2,4-triazole. A library of new indole-3-carbaldehyde-triazole hybrids has been synthesized under conventional and microwave-mediated conditions [34]. The antimicrobial assessment of the compounds showed that 41a–41b, bearing a fluoroquinolone scaffold and 42–44, displayed remarkable activities on Gram-positive and Gram-negative bacteria with MIC values < 0.24 μg/mL. In addition, compounds 41a–41b, 42–44, and 45a–45c showed excellent antifungal activities (see Figure 6).
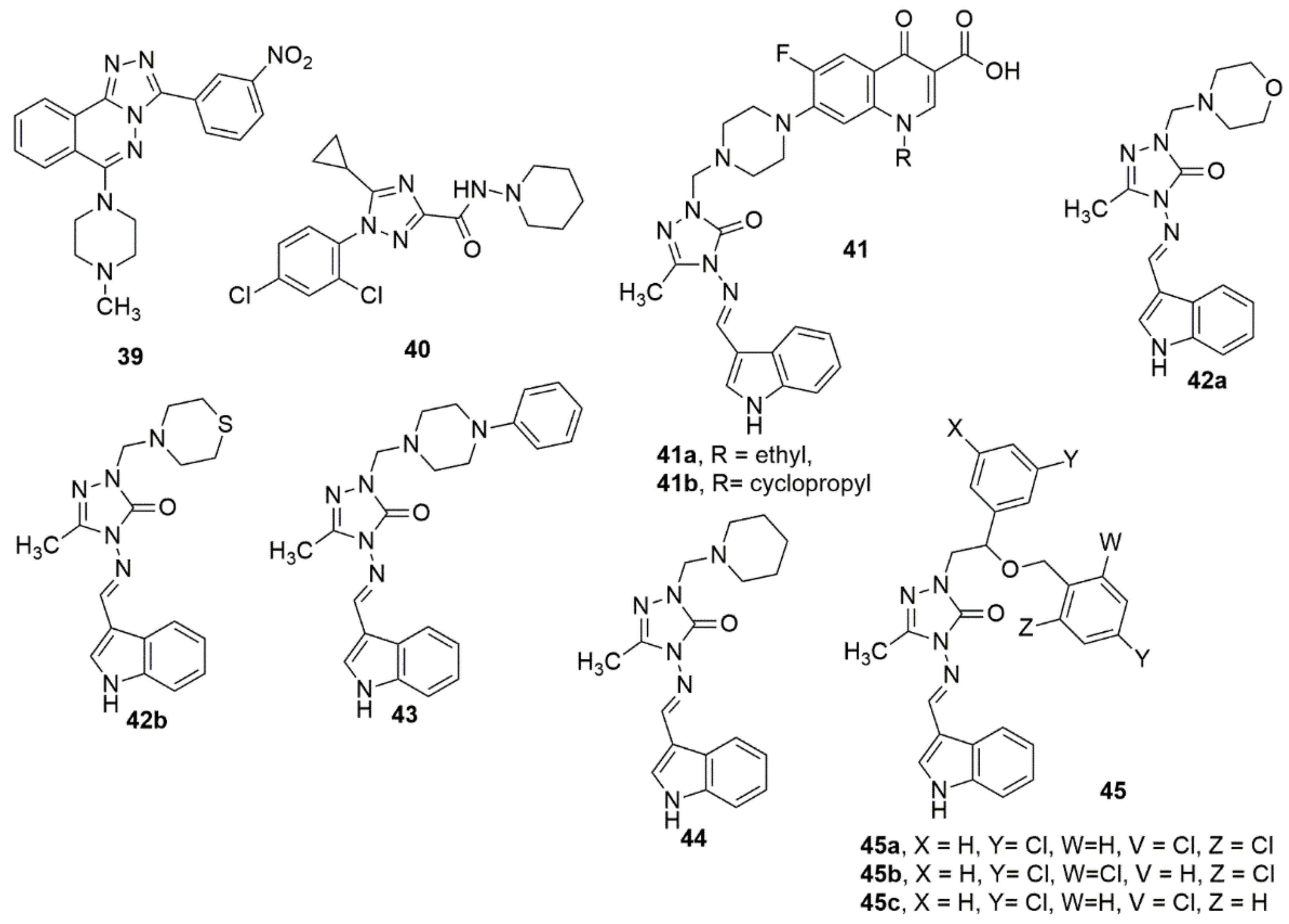
Figure 6. Chemical structures of analogues of 1,2,4-triazole molecule with promising biological activities.
Wang et al. [35] reported a series of interesting compounds and their biological activity evaluation with respect to this tricyclic chemotype of dual PLK1/BRD4 inhibitors to corroborate their effectiveness as anticancer agents. The compounds were synthesized based on the core structure of BI-2536 (PLK1 inhibitor). Among the evaluated compounds, 46 displayed excellent activity for PLK1 (IC50 = 22 nM) and BRD4 (IC50 = 109 nM), with promising antiproliferative activity against a panel of cancer cell lines. Meanwhile, compound 46 displayed equipotent activity with PLK1 (IC50 = 22 nM) and BRD4 (IC50 = 109 nM). The SARS detailed that a bulkier group on the piperazine ring will enhance the stabilized potency between PLK1 and BRD4. Depending on the concentration, the potent compound greatly increased the number of Annexin V/PI-positive MV4-11. This indicates its apoptotic induction effect in cancerous cells, which has also been confirmed by the ascending regulation of apoptosis-associated proteins, including cleaved caspase-3 and cleaved PARP, along with the regulation of the anti-apoptosis protein Bcl-2. In addition, 46 demonstrated favorable in vivo anti-tumor activity with 66% tumor growth inhibition (TGI) at a 60 mg/kg dose without evident toxicity. Wu and coworkers [36] synthesized indole-based [1,2,4]triazolo[4,3-a]pyridine hybrids and screened them for their antiproliferative activities, tubulin polymerization inhibition, and cell cycle arrest/apoptosis-initiating effects. In particular, four cancer cell lines, including human cervical cancer cells (HeLa), human adenocarcinoma epithelial cells (A549), human breast cancer cells (MCF-7), and human colon cancer cells (HCT116), were employed in the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Compound 47 bearing an N-methyl-5-indolyl substituent at the C-6 position of the [1,2,4]triazolo[4,3-a]pyridine moiety displayed superior activity against all the tested cell lines. In addition, compound 47 displayed potent inhibitory activity with respect to tubulin polymerization with an IC50 value of 1.64 ± 0.11 µM, comparable to CA-4 with an IC50 of 1.24 ± 0.08 µM. The primary mechanism of action (MOA) studies demonstrated that 47 could inhibit the proliferative of cancer cells by inducing cell cycle arrest at the G2/M phase and cellular apoptosis in HeLa cells in a dose-dependent manner. The active compound was also detected to have the potential ability to inhibit tumor cell migration and metastasis (see Figure 7).
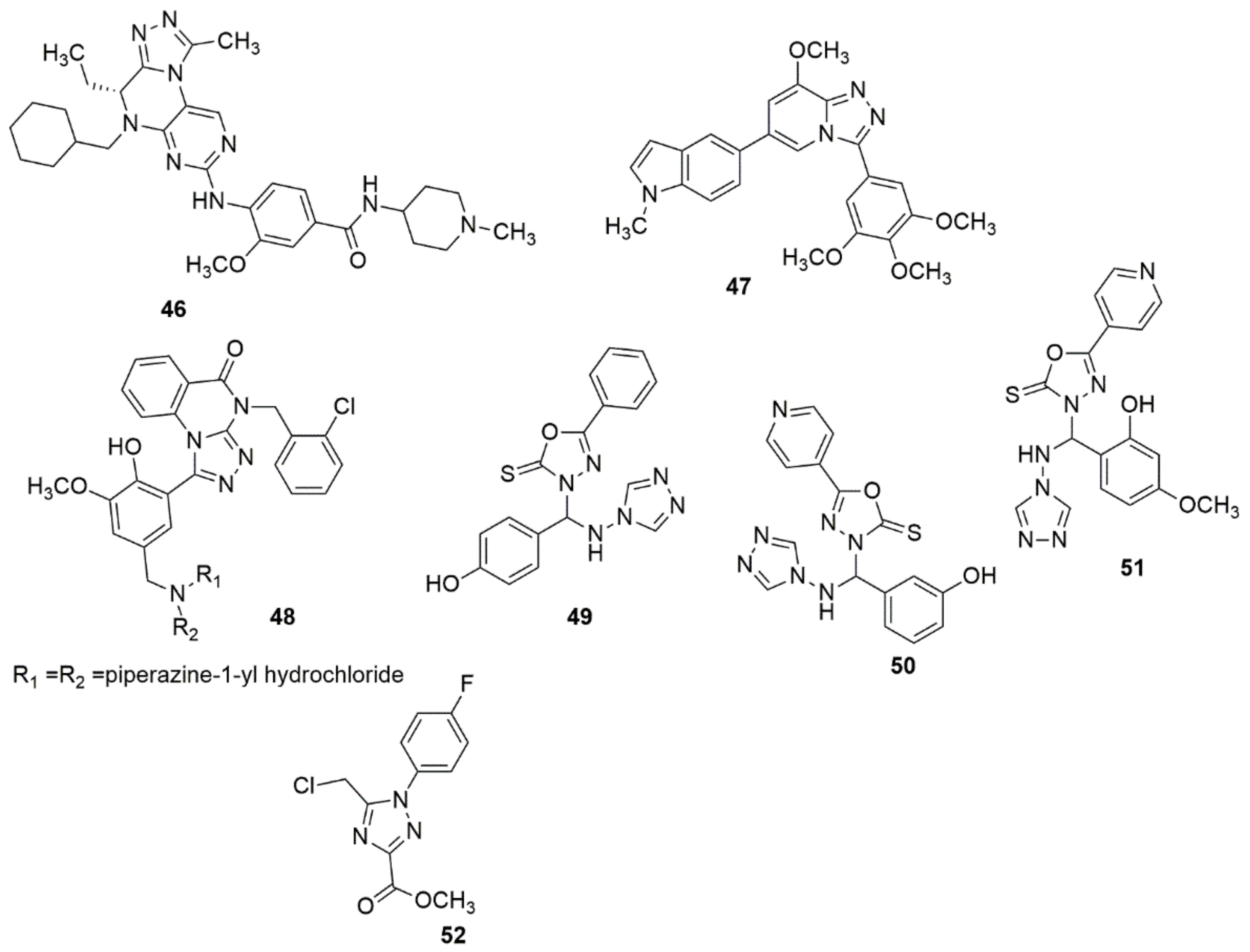
Figure 7. Chemical structures of 1,2,4-triazole hybrids with promising biological activities.
A series of novel triazoloquinazolinone derivatives were designed, synthesized, and evaluated for their inhibitory activity toward the SHP2 protein enzyme [37]. Among the evaluated compounds, 48 displayed the highest inhibitory activity against the SHP2 protein at 10 µM (31.84% inhibition) compared with SHP244. 48 exhibited superior antitumor activities, with an IC50 value of 14.67 µM against A375 cells. The SARs revealed that derivatives with hydroxyl substituents at the two-position of the phenyl ring showed significantly higher activity than derivatives with substituents at the four-positions. In addition, the insertion of electron-withdrawing groups, namely methoxy groups, exhibited enhanced inhibitory activity. Compounds 49–51 have been reported by Jain et al. as being promising for the management of cognitive dysfunction [38]. Li et al. [39] designed and synthesized 1,2,4-triazole-3-carboxylates derivatives; the obtained products were subjected to in vitro NO production and cyclooxygenase COX-1/COX-2 inhibition assays. Notably, compound 52 showed the significant inhibition of NO, COX-2 (IC50 of 2.87 and 17.9 nM), and substantial selectivity (COX-1/COX-2 = 1080). Meanwhile, compound 52 (5 mg/kg) displayed significant in vivo anti-inflammation and gastric protection results, including paw edema, chemokines, and histological experiments, compared to Indomethacin (10 mg/kg). The presence of a fluorine atom enhanced the COX-2 inhibitory activity. The docked complex of 52 showed a comparable interaction landscape with celecoxib in the active COX-2. The active compound formed three hydrogen bond interactions with His75, Leu338, and Phe504 and eight van der Waals interactions with Val335, Leu338, Ser339, Try341, Phe504, Val509, Gly512, and Ala513, respectively (see Figure 7).
A series of novel 3,4,5-trimethoxyphenyl substituted [1,2,4]triazolo[4,3-a]pyridines were designed and synthesized on the basis of triazolopyrimidine 53 as the core compound [40]. The in vitro antiproliferative efficacy of synthesized novel 1,2,4-triazolo[4,3-a]pyridine derivatives were screened against different cancer cell lines using the (MTT) assay. Compound 54 bearing 3-amino-4-methoxyphenyl moiety exhibited the highest activity with an IC50 value of 12 nM, equipotent with CA-4(12nM). Also, 54 was 62-fold superior to compound 53. The active compound 54 also exhibited potent activities against A549, MCF-7, and T47D. The MOA analysis result showed that 54 significantly blocked the cell cycle at the G2/M phase, induced apoptosis in a dose-dependent manner, and disrupted microtubule networks. Compound 54 also exhibited better anti-tubulin activity than CA-4 (see Figure 8).
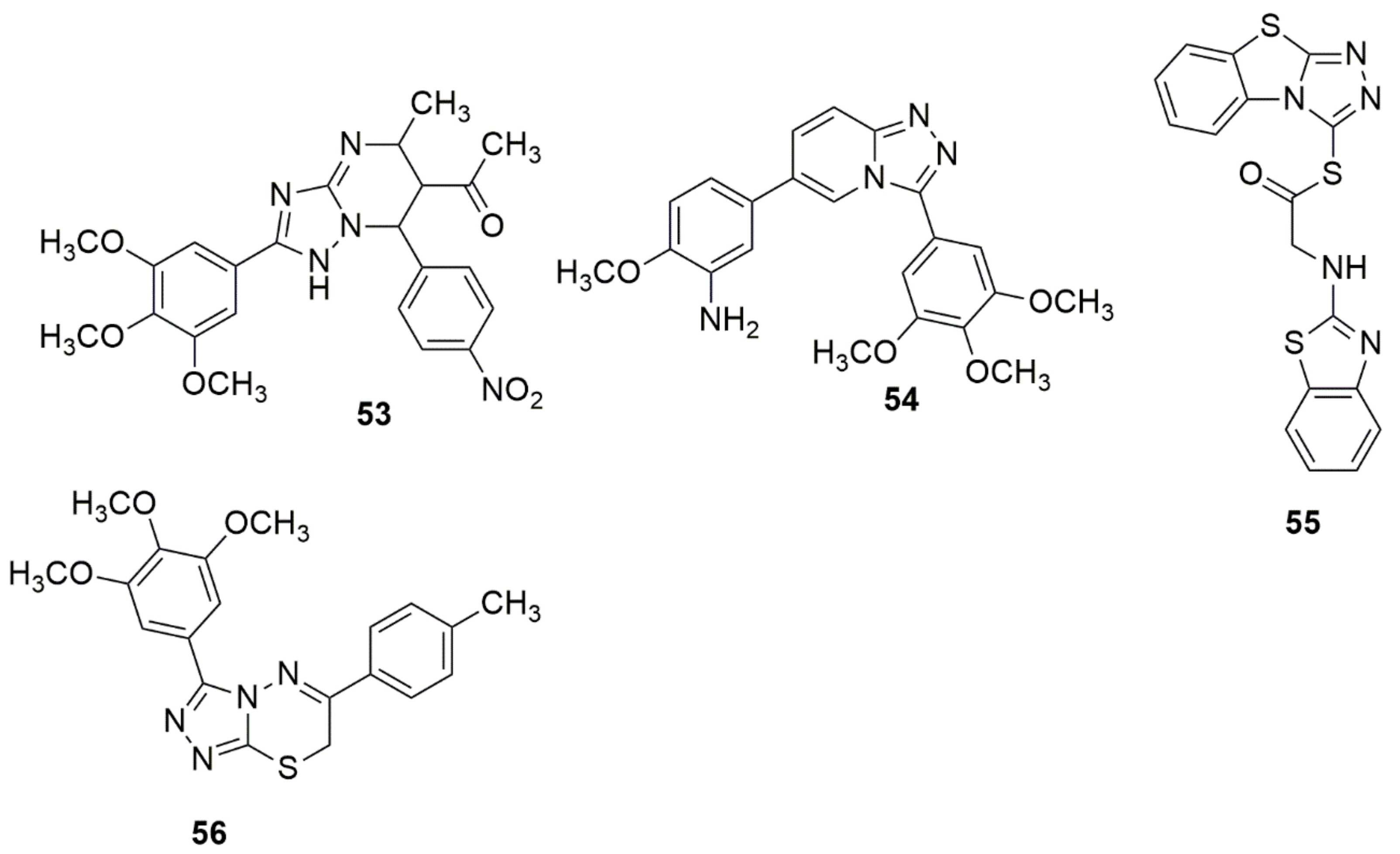
Figure 8. Chemical structures of 1,2,4-triazole hybrids with significant anticancer activities.
Compound 55 has been reported to have substantial apoptosis-inducing activity in A549 cells and inhibited the activity of CDK2/Cyclin A1 with an IC50 value of 4.65 µM [41]. Ma et al. [42] synthesized a novel series of triazolothiadiazine hybrids via the ring-merging approach. The compounds were examined for their in vitro antiproliferative efficacy toward a human colon cancer cell line (HT-29) using an MTT assay. Among the evaluated compounds, 56 demonstrated excellent selectivity over the normal human embryonic kidney HEK-293 cells (IC50 > 100 µM). Compound 56 strongly blocked tubulin polymerization and disrupted intracellular microtubule networks. Compound 56 effectively inhibited the tumor growth of an A549 lung cancer xenograft mouse model without evident signs of toxicity in the in vivo experimentation (see Figure 8).
2.4. Tetrazole
Interest in tetrazole derivatives has increased considerably over the past few decades because of the virtually limitless potential of tetrazole compounds in various fields [43][44][45][46]. They have been successfully applied in pharmaceutical products as a potential replacement for cis-peptide binding. In addition, they are used as components in explosives, ligands in coordination chemistry, and precursors in preparing a diversified selection of heterocyclic compounds [47][48]. Considerable advancement was achieved by Wang et al. [49] by describing the synthesis, antiproliferative, tubulin polymerization, analysis of immunofluorescence staining, and cell cycle analysis of new tetrazole derivatives, 57. Among the compounds synthesized, 57a showed significant activity against SGC-7901, A549, and HeLa cell lines. The SAR detailed that the insertion of substituent into the ortho-position of the ring attached to the nitrogen atom of the triazole ring significantly improved the antiproliferative activity. The compounds bearing 3,4-dimethoxyl showed significant anticancer activities. The tubulin polymerization result showed that 57a disrupts the microtubule network, arrests the cell cycle at the G2/M phase, and induces dose- and time-dependent apoptosis. Ulgheri and co-workers [50] reported designing and synthesizing a new class of active non-peptidomimetic and non-covalent caspase-1 inhibitors. Compound 58a was identified to inhibit IL-1β release in activated macrophages in the low µM range, which corroborates the activities observed for the known covalent inhibitors. Due to the extensive application of altered nucleobases for cancer treatment as a PDE3 inhibitor. Shekouhy et al. [51] presented the synthesis, PDE3 and anticancer properties of some novel nucleobases/tetrazole hybrids using cilostazol as the core structure. Compounds 59a, 59b and 59c are more strong inhibitors of PDE3A than cilostazol, and compound 59b was observed as being the most effective PDE3A inhibitor (see Figure 9).
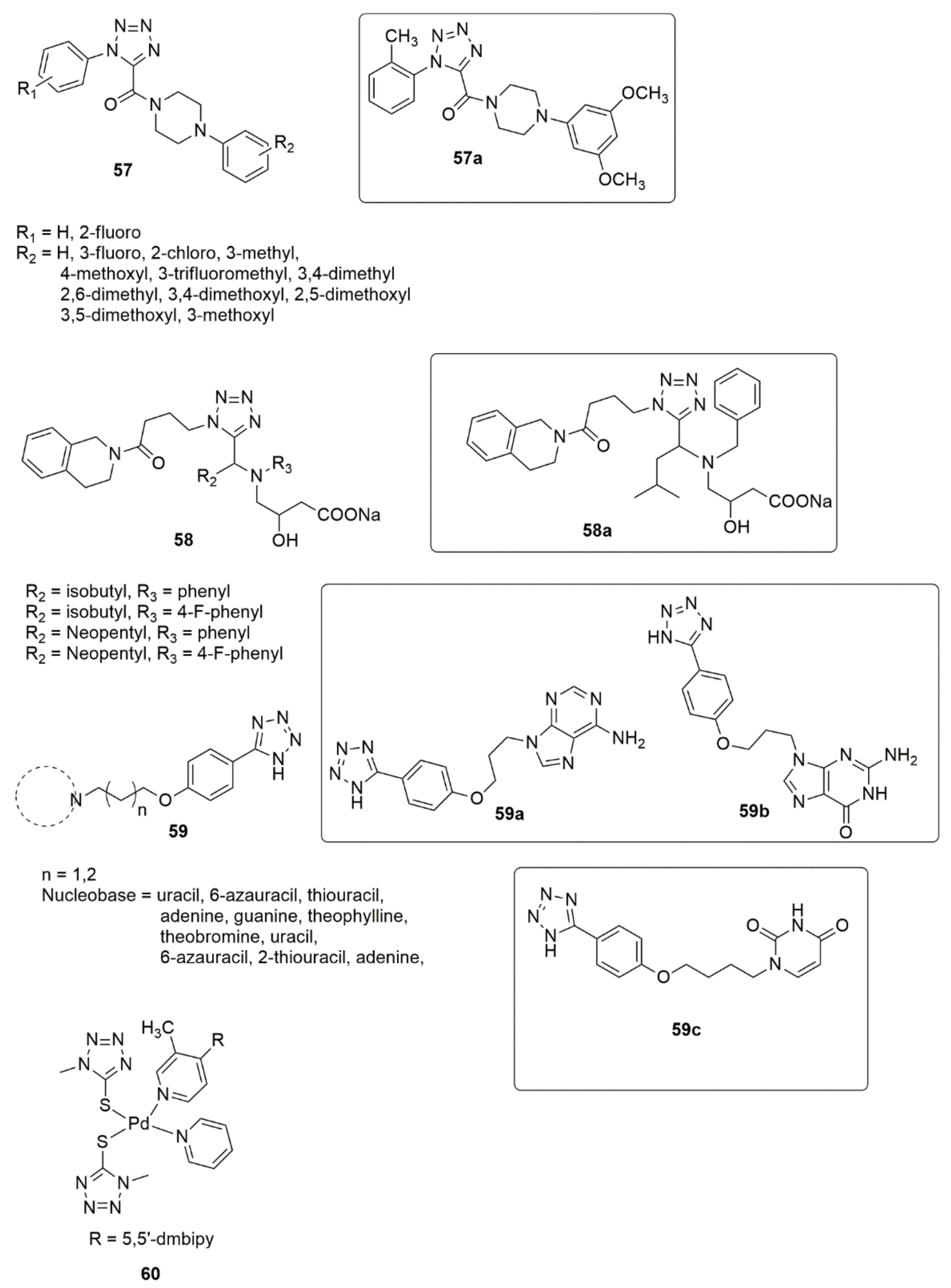
Figure 9. Chemical structures of active tetrazole hybrids with promising anticancer activities.
Additionally, the compounds 59a, 59b and 59c showed significant inhibitory activity against HeLa (IC50 = 27.94 ± 0.36 µM) and MCF-7 (IC50 = 49.22 ± 1.01 μM) cancer cell lines. The presence of two purine-like nucleobases led to the strongest inhibitory effect against the PDE3A, while the insertion of a pyrimidine-like nucleobase also led to an enhanced inhibitory effect against the PDE3A and cytotoxicity activity against the HeLa and MCF-7 cell lines. Rashidipour et al. [52] reported the effectiveness of 60 on SKBR-3 cell proliferation as being similar to that of cisplatin, and the DNA-binding assay uncovered the ability of the compound to bind to DNA and alter its structure (see Figure 9).
2.5. Imidazole/Benzimidazole
A library of new imidazole derivatives 61, 62 have been prepared and evaluated for their biological activity [53]. Most of the examined compounds have significant inhibitory activities. Compound 61a (MIC = 62.5, 100, 100 μg/mL) displayed broad-spectrum antibacterial activity against all ESBL, VRE, and MRSA strains, respectively, while 62a (MIC = 25 μg/mL) showed excellent activity against the ESBL strain. All the examined compounds demonstrated lower activity than the standard drug to inhibit H37Rv strains. The in vitro antimalarial activity against Plasmodium falciparum showed that the analogues 61b (IC50= 0.36 μg/mL), 61a, and 62b (IC50 = 0.45 μg/mL), exhibited moderate activity compared with the reference drug quinine (0.268 μg/mL). Forty novel naphthoquinone phenacylimidazolium derivatives were synthesized and subsequently evaluated for their antitumor activities against three human cancer cell lines [54]. Compound 63 exhibited remarkable activity against the MCF-7 cell line (IC50 = 50 nM) and 256-fold selectivity against normal cells. Furthermore, compound 63 was found to induce apoptosis, activate the pro-apoptotic protein caspase-3, and inhibit survivin expression. Al-Hamashi et al. [55] designed, synthesized, and described a new antimitotic agent class that modulates tubulin polymerization. All the compounds inhibited the growth of HCT 116 cells with GI50 values. The olefin moiety in the linker was crucial for the cytotoxic activity. Hydrogenation of this double bond or conversion to a cyclopropyl moiety obliterated the antiproliferative activity. Substitution of aniline moiety with a cyclohexyl group or a bulky naphthyl moiety lowered the antiproliferative activity, while the substitution of halogen atoms or a trifluoromethyl group at the para-position improved the inhibitory activity. Compound 64a inhibited HDAC1, 2 and 3, while 64a and 64b had no effect on SMC3 acetylation. Compound 64a further destabilized microtubules and accelerated depolymerization. A series of new fluoro-substituted benzimidazole hybrids were designed, synthesized and pharmacologically evaluated [56]. The new compounds were exposed to biological evaluation for their impacts on systolic blood pressure (SBP) and diastolic blood pressure (DBP) in spontaneously hypertensive rats. Of the compounds examined, 65a and 65b reduced blood pressure more effectively and had higher and more enduring antihypertensive effects than losartan and telmisartan at the same dose (see Figure 10).
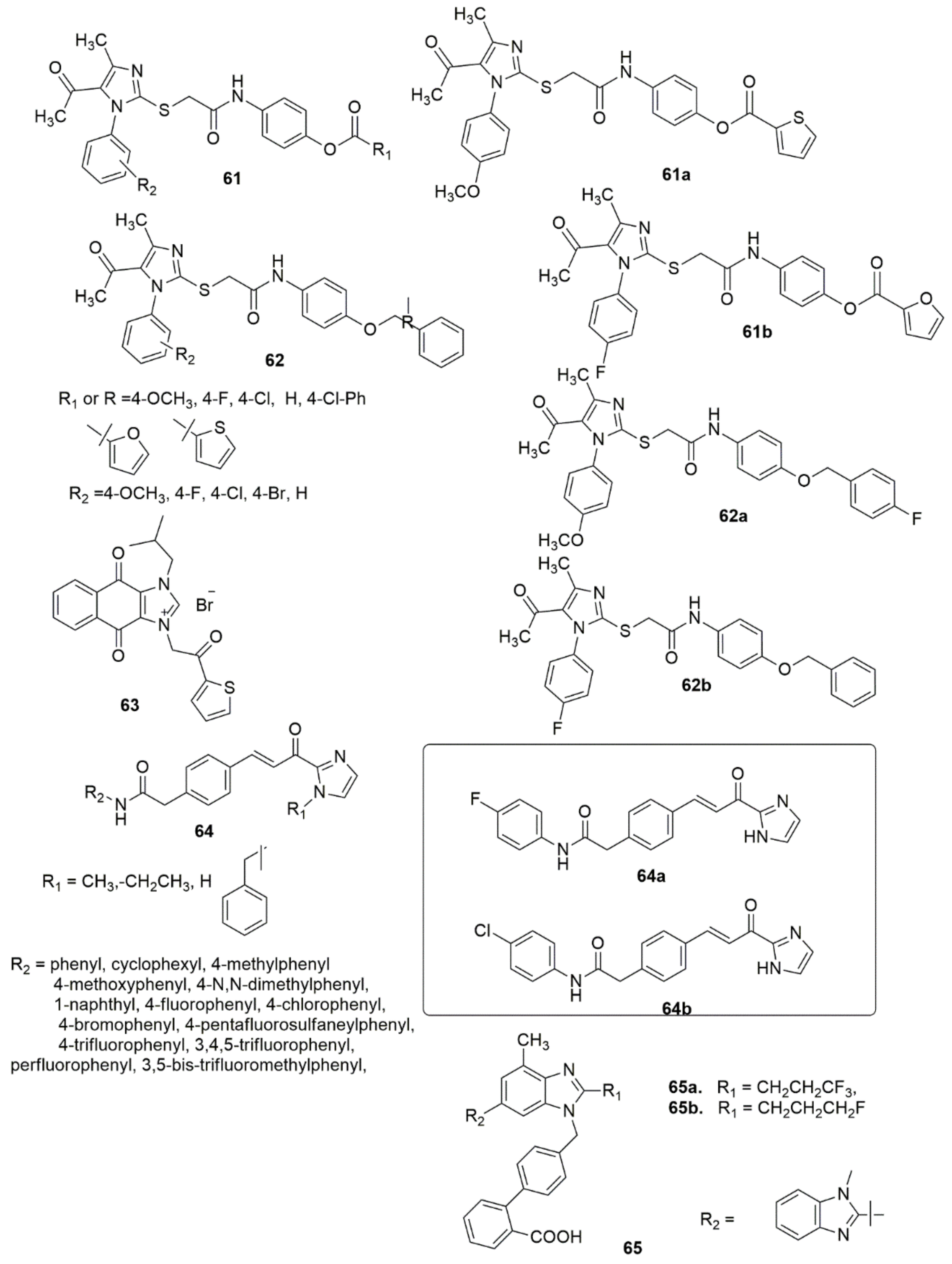
Figure 10. Analogues of imidazole with promising biological activities.
The SARs indicated that the presence of lipophilic benzylamine improved the activity eight-fold in 66b (MIC 1.56 μg/mL), while as a result of the integration of halogens, namely fluorine and chlorine, at the ortho position of the phenyl ring, the inhibitory activity decreased. Among the derivatives with disubstituted halogens, compound 66a (MIC of 0.78 μg/mL) with 3,4-difluoro substituents demonstrated the highest activity. A four-fold reduction in anti-TB activity has been observed with the integration of the electron-withdrawing 4-trifluoromethyl group, whereas the presence of methyl at the ortho position reduced the activity due to the ortho steric clash. However, the activity increased upon incorporating the methoxy group at the same second position. In summary, 66a exhibited the superior anti-tubercular potential with an MIC of 0.78 μg/mL (2.15 μM), followed by 66b, 66c, 66d and 66e with an MIC of 1.56 μg/mL (see Figure 11).
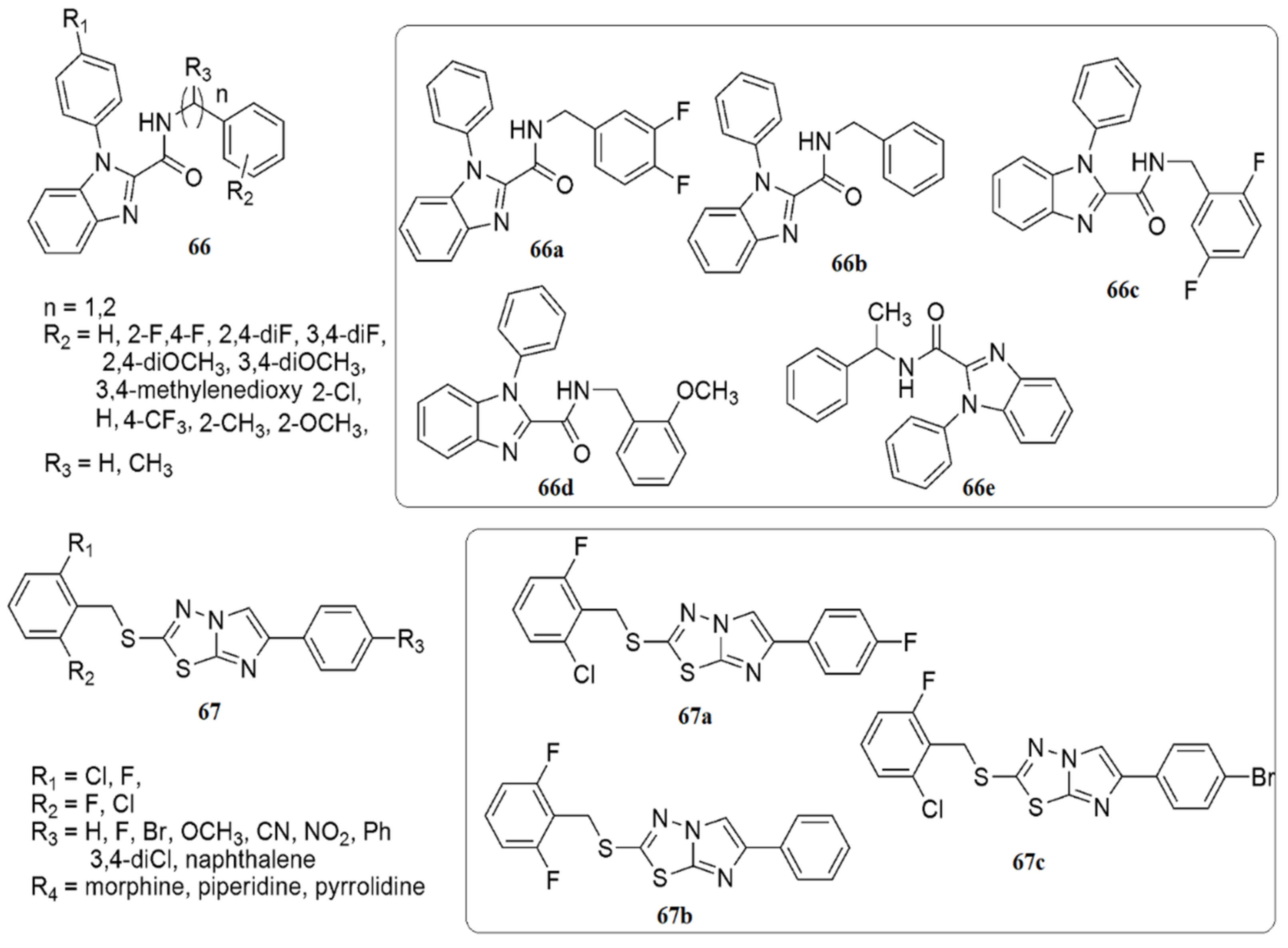
Figure 11. Structures of analogues of imidazole with promising biological activities.
Askin et al. [57] investigated the synthesis, characterization, biological activity, and cytotoxic effects of imidazo[2,1-b][1,3,4]thiadiazole derivatives 67. The novel imidazo[2,1-b][1,3,4]thiadiazole derivatives were tested for their ability to inhibit the ubiquitous cytosolic hCA I and hCA II isozymes and the cholinergic enzyme AChE. All the tested compounds demonstrated low nanomolar inhibitory activity against hCA I, hCA II, and AChE (KIs were 23.44–105.50, 10.32–104.70, and 20.52–54.06 nM, respectively). Moreover, compound 67a inhibits hCA I up to 18-fold compared to acetazolamide, while compound 67b has a five-fold selectivity towards hCA II. 67a, 67b and 67c were the most potent inhibitors of hCA I and II isoforms, AChE, and non-toxic agents against the L929 mouse fibroblast cell line at their effective concentrations on target enzymes (see Figure 11).
Twenty-six novel 4-phenoxypyridine bearing imidazole-4-carboxamide 68 and 4-methyl-5-oxo-4,5-dihydro-1,2,4-triazole-3-carboxamide 69 hybrids were designed, synthesized, and investigated for pharmacological activities [58]. All the newly synthesized target compounds were evaluated for their in vitro inhibitory activity toward c-Met kinase using a mobility shift assay. 69a demonstrated the best activity with an IC50 value of 0.012 μM. The introduction of a fluorine atom on the phenoxy moiety was crucial for c-Met kinase effective activities for the two series of compounds. Based on an antiproliferative assay, compound 69a showed remarkable proliferation reduction effects against MKN-45, A549 and H460 cell lines with IC50 values of 0.64, 1.92 and 2.68 μM, respectively. Additionally, compound 69a strongly inhibited A549 cell motility. The results of a colony formation assay indicated that 69a suppressed the colony formation and prevented the unobstructed increase of A549 cells, and induced apoptosis in MKN-45, A549, and H460 cells, in a concentration-dependent manner (see Figure 12).
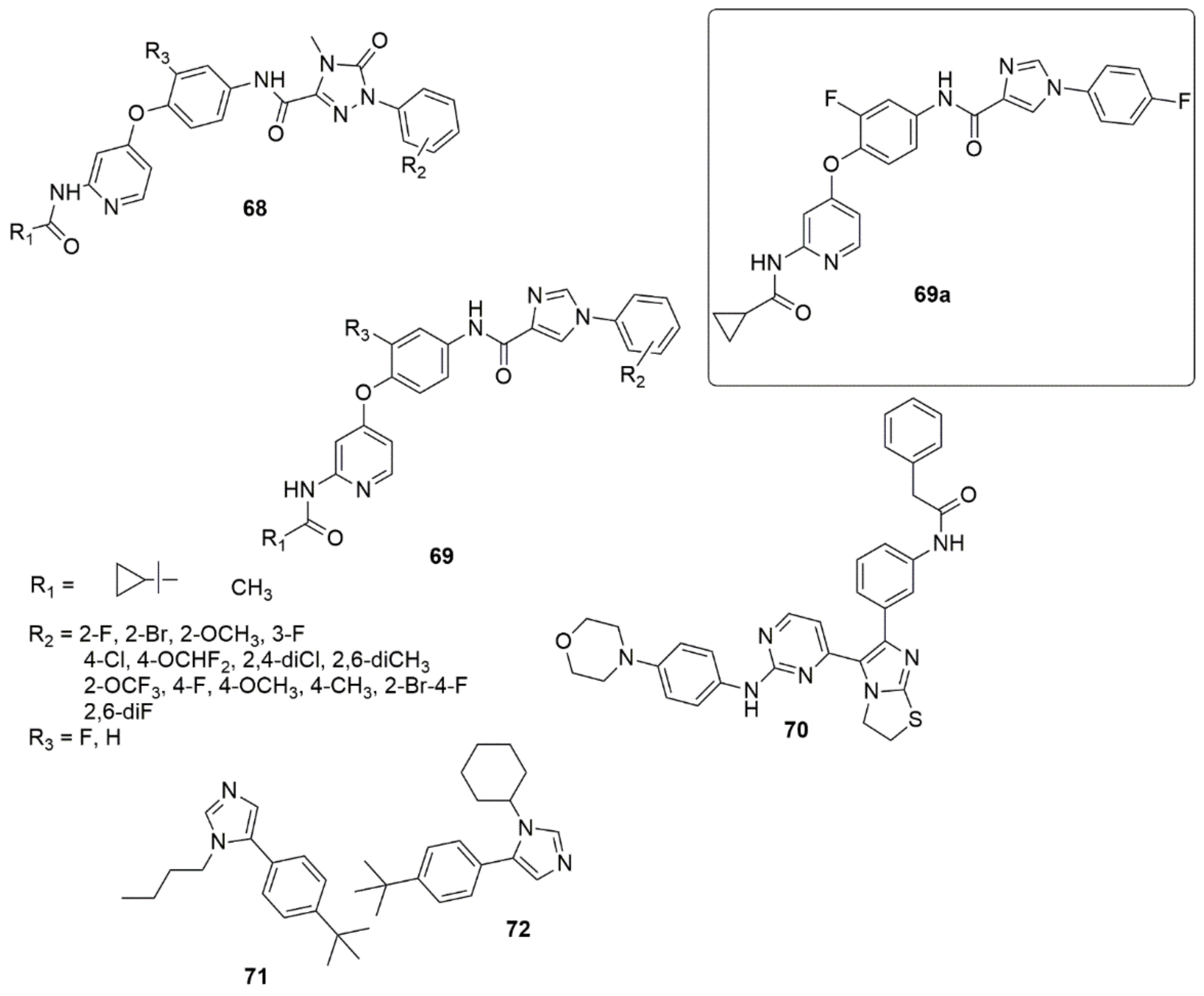
Figure 12. Chemical structures of imidazole hybrids with significant biological activities.
Compound 70 has displayed promising activity with an IC50 value of 52 nM against IGF1R and an IC50 value of 35.5 nM against EGFR with an acceptable PK profile [59]. Compounds 71 and 72 both exhibited activity against HIV-1 in LEDGF/p75 contact, while 71 displayed a MIC value of 15.6 μg/mL against S. aureus, and 72 displayed a comparable MIC value against B. cereus [60] (see Figure 12).
A new library of 2-(5-aryl-1H-imidazol-1-yl) compounds 73, 74, 75 were designed, synthesized, and evaluated for their inhibitory activity against the HIV-1 Vpu and BST-2 protein interaction [61]. The results of the AlphaScreen™ assay showed that 73a and 74b displayed IC50 values of 11.6 ± 1.1, and 17.6 ± 0.9 µM, respectively, in a dose–response profile, whereas in cytotoxicity and antiviral assays, 73a displayed significant activity with an EC50 value of 6.3 ± 0.7 µM at non-toxic concentrations (CC50 = 184.5 ± 0.8 µM), while compound 74b exhibited an EC50 of 157.5 ± 1.2 µM (CC50 = 159.5 ± 0.9 µM). Thus, compound 73a was identified as a potential inhibitor of HIV-1 Vpu and host BST-2 protein. A series of new 3-(4-phenyl-1H-imidazol-2-yl)-1H-pyrazole derivatives were designed and synthesized as JAK 2/3 and Aurora A/B kinase multi-target inhibitors by Zheng et al. [62]. Many of the compounds examined showed good inhibitory activity against JAK2/3 and Aurora A/B (with IC50 values ranging from 0.008 to 2.52 µM). Of all the compounds evaluated, 76a remarkably decreased the toxic effect on normal human cells, more so than JAK 2/3 and the Aurora A/B kinase multi-target kinase inhibitor (AT9832). Compound 76a downregulated the phosphorylation of STAT3, STAT5, Aurora A, and Aurora B in K562 and HCT116 cells. This potent compound induced cell cycle arrest in the G2 phase. Notably, the SAR showed that derivatives bearing a morpholine ring at the side chain exhibited superior antiproliferation activity compared to derivatives bearing a piperidine ring. Additionally, the presence of Cl, OCH3, and NO2 groups at the benzene ring enhanced the proliferative inhibition. However, compounds bearing electron-withdrawing groups such as Cl and NO2 displayed slightly higher K562 proliferative inhibition compared to the compounds containing an electron-donating group (OCH3) (see Figure 13).
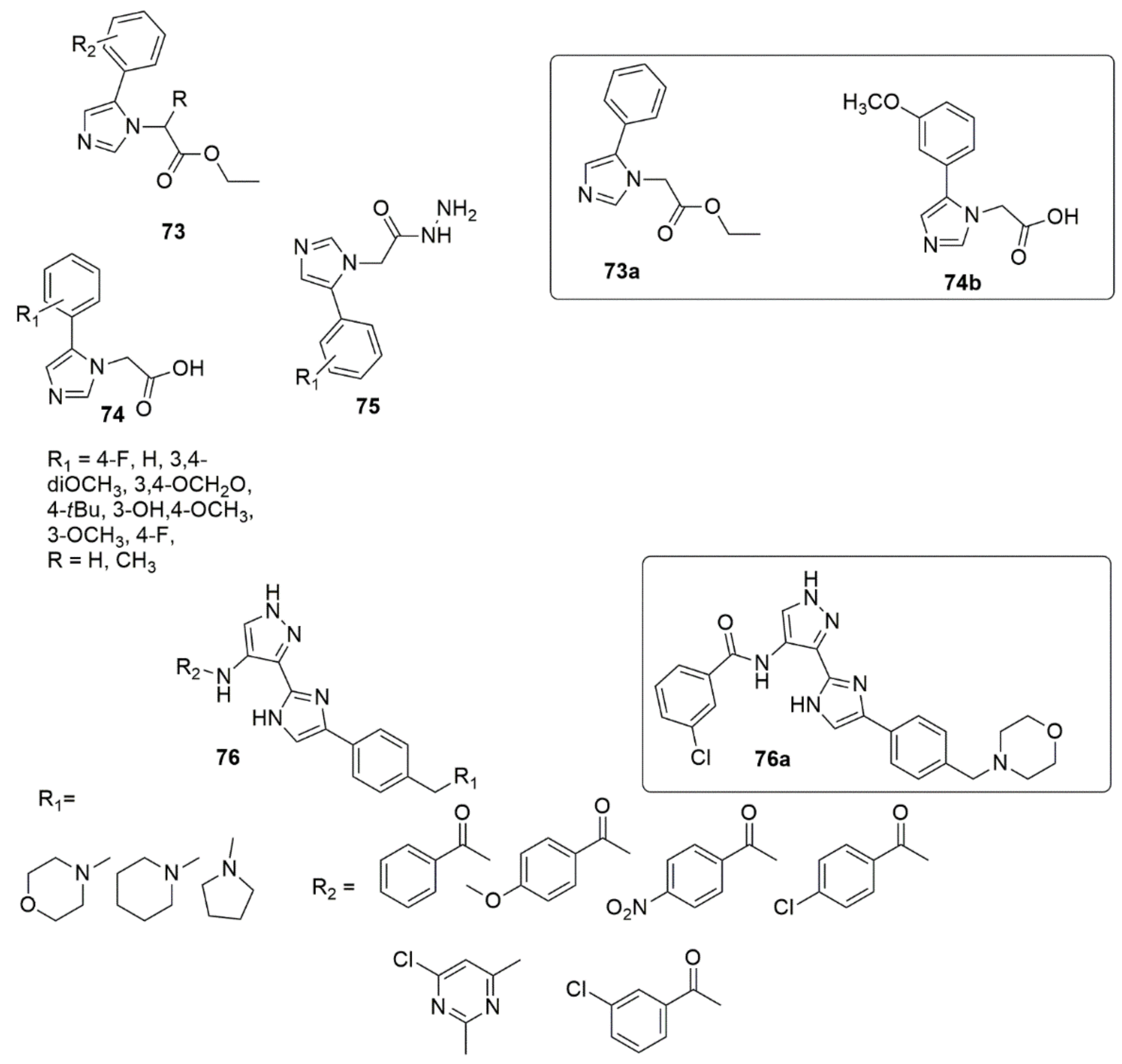
Figure 13. A new library of 2-(5-aryl-1H-imidazol-1-yl) compounds with significant biological activities.
Compound 77 showed potent binding affinity to A2A AR (IC50, 9.2 nM), good selectivity against A1 AR (A2A/A1 80-fold) and high potency in cAMP (IC50 31.0 nM) functional and IL-2 (EC50 = 164.6 nM) production assays. A series of dual imidazole-5-yl pyrimidine inhibitors BRAFV600E/p38a were developed and synthesized to overcome resistance to BRAFV600E inhibitors in BRAFV600E metastatic melanoma patients [63]. Among the examined compounds, 78 exhibited superior dual inhibition with IC50 values of 2.49 and 85 nM against BRAFV600E and p38a, respectively. Compound 78 exhibited high inhibitory activity with an IC50 value of 96.3 nM in the TNF-α production assay. The antiproliferative activity of the targeted compounds was determined using the MTT cytotoxicity assay. Compound 79 showed excellent antiproliferative activity with an IC50 value of 0.9 µM. The compound was 11.11-fold more selective against LOX-IMVI melanoma cells than the IOSE-80PC normal cell line. Lei and co-workers have proposed that 80 could be a potential and promising agent for the treatment of thrombotic diseases [64]. Sekiola et al. [65] discovered compound 81 as an attractive candidate for AD treatments. Compound 117 showed high in vitro potency with brain exposure and displayed an unnoticeable inhibition of cytochrome p450 enzymes. Concentrations of Aß42 in the brain of rats were significantly reduced in vivo at a dose of 10 mg/kg, while the dose of 2 mg/kg of 81 for 8 days fully saved the cognitive deficits of AD model mice. Compound 82 has exhibited excellent inhibitory activities against BRD4(1) with an IC50 value of 0.035 μM [66]. 82 successfully inhibited the proliferation of pancreatic cancer cells BxPC3. Compound 82 also arrested prostate cancer cells in the G0/G1 phase, induced cell apoptosis by regulating the expression of apoptotic proteins and demonstrated effective in vivo antitumor activity by inducing the apoptosis of tumor cells. Bu et al. [67] proposed compound 83 as potential MNK1/2 inhibitor [67]. Compound 84 has been shown to be a potential starting point for the development of a lead molecule used for the treatment of leukemia and glioblastoma (see Figure 14).
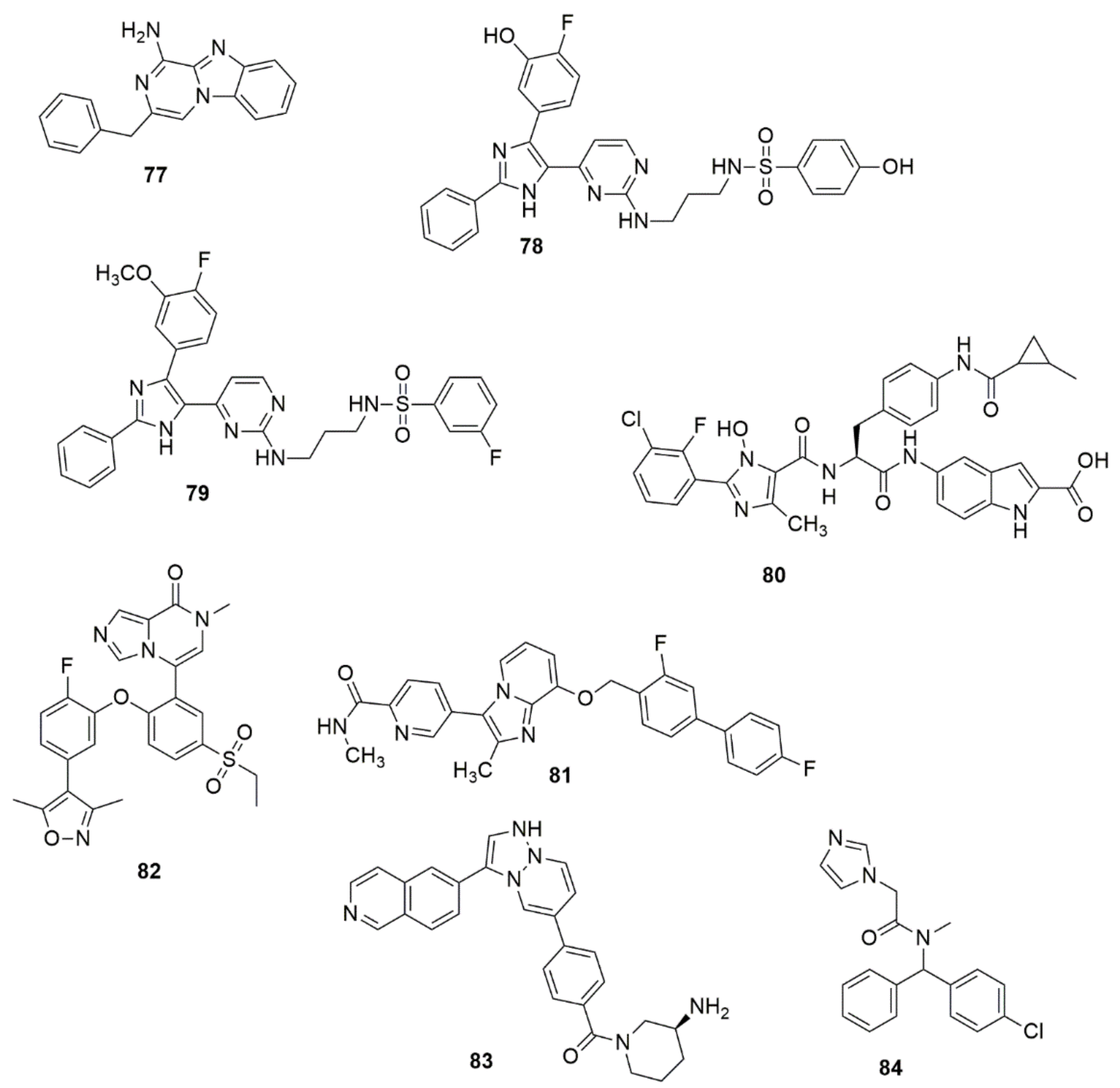
Figure 14. Structures of analogues of imidazole molecule with promising biological activities.
References
- Li, W.T.; Hwang, D.R.; Chen, C.P.; Shen, C.W.; Huang, C.L.; Chen, T.W.; Lin, C.H.; Chang, Y.L.; Chang, Y.Y.; Lo, Y.K.; et al. Synthesis and biological evaluation of N-heterocyclic indolyl glyoxylamides as orally active anticancer agents. J. Med. Chem. 2003, 46, 1706–1715.
- Sörgel, F.; Kinzig, M. Pharmacokinetics of gyrase inhibitors, Part 1: Basic chemistry and gastrointestinal disposition. Am. J. Med. 1993, 94, 44–55.
- Zhou, C.H.; Wang, Y. Recent researches in triazole compounds as medicinal drugs. Curr. Med. Chem. 2012, 19, 239–280.
- Riu, F.; Sanna, L.; Ibba, R.; Piras, S.; Bordoni, V.; Scorciapino, M.A.; Lai, M.; Sestito, S.; Bagella, L.; Carta, A. A comprehensive assessment of a new series of 5′,6′-difluorobenzotriazole-acrylonitrile derivatives as microtubule targeting agents (MTAs). Eur. J. Med. Chem. 2021, 222, 113590.
- Kasemsuk, T.; Saehlim, N.; Arsakhant, P.; Sittithumcharee, G.; Okada, S.; Saeeng, R. A novel synthetic acanthoic acid analogues and their cytotoxic activity in cholangiocarcinoma cells. Bioorg. Med. Chem. 2021, 29, 115886.
- Felipe, J.L.; Cassamale, T.B.; Lourenço, L.D.; Carvalho, D.B.; das Neves, A.R.; Duarte, R.C.F.; Carvalho, M.G.; Toffoli-Kadri, M.C.; Baroni, A.C.M. Anti-inflammatory, ulcerogenic and platelet activation evaluation of novel 1,4-diaryl-1,2,3-triazole neolignan-celecoxib hybrids. Bioorg. Chem. 2022, 119, 105485.
- Holanda, V.N.; da Silva, W.V.; do Nascimento, P.H.; Silva, S.R.B.; Cabral Filho, P.E.; de Oliveira Assis, S.P.; da Silva, C.A.; de Oliveira, R.N.; de Figueiredo, R.C.B.Q.; de Menezes Lima, V.L. Antileishmanial activity of 4-phenyl-1--1H-1, 2, 3-triazole (PT4) derivative on Leishmania amazonensis and Leishmania braziliensis: In silico ADMET, in vitro activity, docking and molecular dynamic simulations. Bioorg. Chem. 2020, 105, 104437.
- Chu-Moyer, M.Y.; Ballinger, W.E.; Beebe, D.A.; Berger, R.; Coutcher, J.B.; Day, W.W.; Li, J.; Mylari, B.L.; Oates, P.J.; Weekly, R.M. Orally-effective, long-acting sorbitol dehydrogenase inhibitors: Synthesis, structure-activity relationships, and in vivo evaluations of novel heterocycle-substituted piperazino-pyrimidines. J. Med. Chem. 2002, 45, 511–528.
- Agrahari, A.K.; Singh, A.K.; Singh, A.S.; Singh, M.; Maji, P.; Yadav, S.; Rajkhowa, S.; Prakash, P.; Tiwari, V.K. Click inspired synthesis of p-tert-butyl calix arene tethered benzotriazolyl dendrimers and their evaluation as anti-bacterial and anti-biofilm agents. New J. Chem. 2020, 44, 19300–19313.
- El Malah, T.; Mageid, R.E.A.; Awad, H.M.; Nour, H.F. Copper (i)-catalysed azide–alkyne cycloaddition and antiproliferative activity of mono-and bis-1, 2, 3-triazole derivatives. New J. Chem. 2020, 44, 18256–18263.
- Şahin, İ.; Çeşme, M.; Özgeriş, F.B.; Güngör, Ö.; Tümer, F. Design and synthesis of 1,4-disubstituted 1,2,3-triazoles: Biological evaluation, in silico molecular docking and ADME screening. J. Mol. Struct. 2022, 1247, 131344.
- Zhang, G.-Y.; Zhang, Z.; Li, K.; Liu, J.; Li, B.; Jin, Z.; Liu, Y.-H.; Tang, Y.-Z. Design, synthesis and biological evaluation of novel pleuromutilin derivatives containing piperazine and 1, 2, 3-triazole linker. Bioorg. Chem. 2020, 105, 104398.
- Hao, S.; Cheng, X.; Wang, X.; An, R.; Xu, H.; Guo, M.; Li, C.; Wang, Y.; Hou, Z.; Guo, C. Design, synthesis and biological evaluation of novel carbohydrate-based sulfonamide derivatives as antitumor agents. Bioorg. Chem. 2020, 104, 104237.
- Pal, T.; Bhimaneni, S.; Sharma, A.; Flora, S. Design, synthesis, biological evaluation and molecular docking study of novel pyridoxine–triazoles as anti-Alzheimer’s agents. RSC Adv. 2020, 10, 26006–26021.
- Aneja, B.; Queen, A.; Khan, P.; Shamsi, F.; Hussain, A.; Hasan, P.; Rizvi, M.M.A.; Daniliuc, C.G.; Alajmi, M.F.; Mohsin, M. Design, synthesis & biological evaluation of ferulic acid-based small molecule inhibitors against tumor-associated carbonic anhydrase IX. Bioorg. Med. Chem. 2020, 28, 115424.
- Suryanarayana, K.; Maddila, S.; Nagaraju, K.; Jonnalagadda, S.B. Design, synthesis, docking study and biological evaluation of novel thieno-pyrimidine tethered 1,2,3-triazole scaffolds. J. Mol. Struct. 2022, 1250, 131713.
- Nemati, F.; Salehi, P.; Bararjanian, M.; Hadian, N.; Mohebbi, M.; Lauro, G.; Ruggiero, D.; Terracciano, S.; Bifulco, G.; Bruno, I. Discovery of noscapine derivatives as potential β-tubulin inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 127489.
- Sepehri, N.; Asemanipoor, N.; Mousavianfard, S.A.; Hoseini, S.; Faramarzi, M.A.; Adib, M.; Biglar, M.; Larijani, B.; Hamedifar, H.; Mohammadi-Khanaposhtani, M. New acridine-9-carboxamide linked to 1, 2, 3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: Design, synthesis, in vitro, and in silico biological evaluations. Med. Chem. Res. 2020, 29, 1836–1845.
- Cherif, M.; Horchani, M.; Al-Ghamdi, Y.O.; Almalki, S.G.; Alqurashi, Y.E.; Jannet, H.B.; Romdhane, A. New pyrano-1, 2, 3-triazolopyrimidinone derivatives as anticholinesterase and antibacterial agents: Design, microwave-assisted synthesis and molecular docking study. J. Mol. Struct. 2020, 1220, 128685.
- Sun, P.; Zhu, Y.; Han, Y.; Hu, K.; Huang, S.; Wang, M.; Wu, H.; Tang, G. Radiosynthesis and biological evaluation of an fluorine-18 labeled galactose derivative FPGal for imaging the hepatic asialoglycoprotein receptor. Bioorg. Med. Chem. Lett. 2020, 30, 127187.
- Shi, S.; Wang, H.; Wang, J.; Wang, Y.; Xue, X.; Hou, Z.; Yao, G.-D.; Huang, X.-X.; Zhao, H.; Liu, Q. Semi-synthesis and biological evaluation of flavone hybrids as multifunctional agents for the potential treatment of Alzheimer’s disease. Bioorg. Chem. 2020, 100, 103917.
- Tangadanchu, V.K.R.; Jiang, H.; Yu, Y.; Graham, T.J.; Liu, H.; Rogers, B.E.; Gropler, R.; Perlmutter, J.; Tu, Z. Structure-activity relationship studies and bioactivity evaluation of 1, 2, 3-triazole containing analogues as a selective sphingosine kinase-2 inhibitors. Eur. J. Med. Chem. 2020, 206, 112713.
- Feng, Y.; Wang, W.; Zhang, Y.; Fu, X.; Ping, K.; Zhao, J.; Lei, Y.; Mou, Y.; Wang, S. Synthesis and biological evaluation of celastrol derivatives as potential anti-glioma agents by activating RIP1/RIP3/MLKL pathway to induce necroptosis. Eur. J. Med. Chem. 2022, 229, 114070.
- Chaidam, S.; Saehlim, N.; Athipornchai, A.; Sirion, U.; Saeeng, R. Synthesis and biological evaluation of 1, 6-bis-triazole-2, 3, 4-tri-O-benzyl-α-d-glucopyranosides as a novel α-glucosidase inhibitor in the treatment of Type 2 diabetes. Bioorg. Med. Chem. Lett. 2021, 50, 128331.
- Payne, M.; Bottomley, A.L.; Och, A.; Hiscocks, H.G.; Asmara, A.P.; Harry, E.J.; Ung, A.T. Synthesis and biological evaluation of tetrahydroisoquinoline-derived antibacterial compounds. Bioorg. Med. Chem. 2022, 57, 116648.
- Le-Nhat-Thuy, G.; Thi, N.N.; Pham-The, H.; Thi, T.A.D.; Thi, H.N.; Thi, T.H.N.; Hoang, S.N.; Van Nguyen, T. Synthesis and biological evaluation of novel quinazoline-triazole hybrid compounds with potential use in Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2020, 30, 127404.
- Begam, R.; Shajahan, A.; Shefin, B.; Murugan, V. Synthesis of novel naphthalimide tethered 1,2,3-triazoles: In vitro biological evaluation and docking study of anti-inflammatory inhibitors. J. Mol. Struct. 2022, 1254, 132364.
- Hosseini, S.; Pourmousavi, S.A.; Mahdavi, M.; Taslimi, P. Synthesis, and in vitro biological evaluations of novel naphthoquinone conjugated to aryl triazole acetamide derivatives as potential anti-Alzheimer agents. J. Mol. Struct. 2022, 1255, 132229.
- Gurrapu, N.; Kumar, E.P.; Kolluri, P.K.; Putta, S.; Sivan, S.K.; Subhashini, N. Synthesis, biological evaluation and molecular docking studies of novel 1, 2, 3-triazole tethered chalcone hybrids as potential anticancer agents. J. Mol. Struct. 2020, 1217, 128356.
- Abdel-Hafez, G.A.; Mohamed, A.-M.I.; Youssef, A.F.; Simons, C.; Aboraia, A.S. Synthesis, computational study and biological evaluation of 9-acridinyl and 1-coumarinyl-1, 2, 3-triazole-4-yl derivatives as topoisomerase II inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 502–513.
- Vo, D.V.; Hong, K.H.; Lee, J.; Park, H. Synthesis, in vitro evaluation, and computational simulations studies of 1,2,3-triazole analogues as DPP-4 inhibitors. Bioorg. Med. Chem. 2021, 29, 115861.
- Romero, A.H.; Sojo, F.; Arvelo, F.; Calderón, C.; Morales, A.; López, S.E. Anticancer potential of new 3-nitroaryl-6-(N-methyl) piperazin-1, 2, 4-triazolo phthalazines targeting voltage-gated K+ channel: Copper-catalyzed one-pot synthesis from 4-chloro-1-phthalazinyl-arylhydrazones. Bioorg. Chem. 2020, 101, 104031.
- Tsai, S.-E.; Li, S.-M.; Tseng, C.-C.; Chung, C.-Y.; Zeng, Y.-H.; Lin, C.C.; Fuh, M.-T.; Yang, L.-C.; Yang, Y.-C.; Wong, F.-F. Chlorotrimethylsilane promoted one-flask heterocyclic synthesis of 1, 2, 4-triazoles from nitrilimines: Modeling studies and bioactivity evaluation of LH-21 and Rimonabant analogues. Bioorg. Chem. 2020, 104, 104299.
- Cebeci, Y.U.; Ceylan, Ş.; Karaoğlu, Ş.A. Conventional and microwave irradiated synthesis, biological activity evaluation of highly substituted indole-triazole hybrids. J. Mol. Struct. 2022, 1250, 131799.
- Wang, N.-Y.; Xu, Y.; Xiao, K.-J.; Zuo, W.-Q.; Zhu, Y.-X.; Hu, R.; Wang, W.-L.; Shi, Y.-J.; Yu, L.-T.; Liu, Z.-H. Design, synthesis, and biological evaluation of 4, 5-dihydro- triazolo pteridine derivatives as novel dual-PLK1/BRD4 inhibitors. Eur. J. Med. Chem. 2020, 191, 112152.
- Wu, C.J.; Wu, J.Q.; Hu, Y.; Pu, S.; Lin, Y.; Zeng, Z.; Hu, J.; Chen, W.H. Design, synthesis and biological evaluation of indole-based triazolo pyridine derivatives as novel microtubule polymerization inhibitors. Eur. J. Med. Chem. 2021, 223, 113629.
- Luo, R.; Wang, Z.; Luo, D.; Qin, Y.; Zhao, C.; Yang, D.; Lu, T.; Zhou, Z.; Huang, Z. Design, synthesis, and biological evaluation of novel triazoloquinazolinone derivatives as SHP2 protein inhibitors. J. Enzym. Inhib. Med. Chem. 2021, 36, 2170–2182.
- Jain, A.; Piplani, P. Design, synthesis and biological evaluation of triazole-oxadiazole conjugates for the management of cognitive dysfunction. Bioorg. Chem. 2020, 103, 104151.
- Li, S.-M.; Tsai, S.-E.; Chiang, C.-Y.; Chung, C.-Y.; Chuang, T.-J.; Tseng, C.-C.; Jiang, W.-P.; Huang, G.-J.; Lin, C.-Y.; Yang, Y.-C. New methyl 5-(halomethyl)-1-aryl-1H-1, 2, 4-triazole-3-carboxylates as selective COX-2 inhibitors and anti-inflammatory agents: Design, synthesis, biological evaluation, and docking study. Bioorg. Chem. 2020, 104, 104333.
- Yang, F.; Jian, X.-E.; Diao, P.-C.; Huo, X.-S.; You, W.-W.; Zhao, P.-L. Synthesis, and biological evaluation of 3, 6-diaryl- triazolo pyridine analogues as new potent tubulin polymerization inhibitors. Eur. J. Med. Chem. 2020, 204, 112625.
- Abdelazeem, A.H.; Alqahtani, A.M.; Omar, H.A.; Bukhari, S.N.A.; Gouda, A.M. Synthesis, biological evaluation and kinase profiling of novel S-benzo thiazolo triazole derivatives as cytotoxic agents with apoptosis-inducing activity. J. Mol. Struct. 2020, 1219, 128567.
- Ma, W.; Chen, P.; Huo, X.; Ma, Y.; Li, Y.; Diao, P.; Yang, F.; Zheng, S.; Hu, M.; You, W. Development of triazolothiadiazine derivatives as highly potent tubulin polymerization inhibitors: Structure-activity relationship, in vitro and in vivo study. Eur. J. Med. Chem. 2020, 208, 112847.
- Wittenberger, S.J. Recent developments in tetrazole chemistry. A review. Org. Prep. Proced. Int. 1994, 26, 499–531.
- Myznikov, L.; Hrabalek, A.; Koldobskii, G. Drugs in the tetrazole series. Chem. Heterocycl. Compd. 2007, 43, 1–9.
- Zhao, H.; Qu, Z.-R.; Ye, H.-Y.; Xiong, R.-G. In situ hydrothermal synthesis of tetrazole coordination polymers with interesting physical properties. Chem. Soc. Rev. 2008, 37, 84–100.
- Yoneyama, H.; Usami, Y.; Komeda, S.; Harusawa, S. Efficient transformation of inactive nitriles into 5-substituted 1H-tetrazoles using microwave irradiation and their applications. Synthesis 2013, 45, 1051–1059.
- Herr, R.J. 5-Substituted-1H-tetrazoles as carboxylic acid isosteres: Medicinal chemistry and synthetic methods. Bioorg. Med. Chem. 2002, 10, 3379–3393.
- Wei, C.X.; Bian, M.; Gong, G.H. Tetrazolium compounds: Synthesis and applications in medicine. Molecules 2015, 20, 5528–5553.
- Wang, C.; Li, Y.; Liu, Z.; Wang, Z.; Liu, Z.; Man, S.; Zhang, Y.; Bao, K.; Wu, Y.; Guan, Q.; et al. Design, synthesis and biological evaluation of 1-Aryl-5-(4-arylpiperazine-1-carbonyl)-1H-tetrazols as novel microtubule destabilizers. J. Enzym. Inhib. Med. Chem. 2021, 36, 549–560.
- Ulgheri, F.; Spanu, P.; Deligia, F.; Loriga, G.; Fuggetta, M.P.; de Haan, I.; Chandgudge, A.; Groves, M.; Domling, A. Design, synthesis and biological evaluation of 1,5-disubstituted α-amino tetrazole derivatives as non-covalent inflammasome-caspase-1 complex inhibitors with potential application against immune and inflammatory disorders. Eur. J. Med. Chem. 2022, 229, 114002.
- Shekouhy, M.; Karimian, S.; Moaddeli, A.; Faghih, Z.; Delshad, Y.; Khalafi-Nezhad, A. The synthesis and biological evaluation of nucleobases/tetrazole hybrid compounds: A new class of phosphodiesterase type 3 (PDE3) inhibitors. Bioorg. Med. Chem. 2020, 28, 115540.
- Rashidipour, A.; Alizadeh, R.; Sadeghi Mohammadi, S.; Tohidlou, M.; Amani, V.; Seyfi, S. Synthesis, crystal structures and biological activity of palladium(II) complexes with 1-methyl-1H-1,2,3,4-tetrazole-5-thiol and substituted 2,2′-bipyridines. J. Coord. Chem. 2020, 73, 3249–3266.
- Daraji, D.G.; Rajani, D.P.; Rajani, S.D.; Pithawala, E.A.; Jayanthi, S.; Patel, H.D. Structure based design, synthesis, and biological evaluation of imidazole derivatives targeting dihydropteroate synthase enzyme. Bioorg. Med. Chem. Lett. 2021, 36, 127819.
- Yuan, J.; Liu, Z.; Zhang, Z.; Yan, D.; Zhang, W. Synthesis and biological evaluation of naphthoquinone phenacylimidazolium derivatives. Bioorg. Med. Chem. Lett. 2021, 41, 127977.
- Al-Hamashi, A.A.; Koranne, R.; Dlamini, S.; Alqahtani, A.; Karaj, E.; Rashid, M.S.; Knoff, J.R.; Dunworth, M.; Pflum, M.K.H.; Casero, R.A.; et al. A new class of cytotoxic agents targets tubulin and disrupts microtubule dynamics. Bioorg. Chem. 2021, 116, 105297.
- Wu, Z.; Xia, M.B.; Bertsetseg, D.; Wang, Y.H.; Bao, X.L.; Zhu, W.B.; Tao, X.; Chen, P.R.; Tang, H.S.; Yan, Y.J.; et al. Design, synthesis and biological evaluation of novel fluoro-substituted benzimidazole derivatives with anti-hypertension activities. Bioorg. Chem. 2020, 101, 104042.
- Askin, S.; Tahtaci, H.; Türkeş, C.; Demir, Y.; Ece, A.; Akalın Çiftçi, G.; Beydemir, Ş. Design, synthesis, characterization, in vitro and in silico evaluation of novel imidazothiadiazoles as highly potent acetylcholinesterase and non-classical carbonic anhydrase inhibitors. Bioorg. Chem 2021, 113, 105009.
- Liu, J.; Liu, F.; Li, Z.; Li, C.; Wu, S.; Shen, J.; Wang, H.; Du, S.; Wei, H.; Hou, Y.; et al. Novel 4-phenoxypyridine derivatives bearing imidazole-4-carboxamide and 1,2,4-triazole-3-carboxamide moieties: Design, synthesis and biological evaluation as potent antitumor agents. Bioorg. Chem. 2022, 120, 105629.
- Gadekar, P.K.; Urunkar, G.; Roychowdhury, A.; Sharma, R.; Bose, J.; Khanna, S.; Damre, A.; Sarveswari, S. Design, synthesis and biological evaluation of 2,3-dihydroimidazothiazoles as dual EGFR and IGF1R inhibitors. Bioorg. Chem. 2021, 115, 105151.
- Rashamuse, T.J.; Harrison, A.T.; Mosebi, S.; van Vuuren, S.; Coyanis, E.M.; Bode, M.L. Design, synthesis and biological evaluation of imidazole and oxazole fragments as HIV-1 integrase-LEDGF/p75 disruptors and inhibitors of microbial pathogens. Bioorg. Med. Chem. 2020, 28, 115210.
- Rashamuse, T.J.; Njengele, Z.; Coyanis, E.M.; Sayed, Y.; Mosebi, S.; Bode, M.L. Design, synthesis and biological evaluation of novel 2-(5-aryl-1H-imidazol-1-yl) derivatives as potential inhibitors of the HIV-1 Vpu and host BST-2 protein interaction. Eur. J. Med. Chem. 2020, 190, 112111.
- Zheng, Y.G.; Wang, J.A.; Meng, L.; Pei, X.; Zhang, L.; An, L.; Li, C.L.; Miao, Y.L. Design, synthesis, biological activity evaluation of 3-(4-phenyl-1H-imidazol-2-yl)-1H-pyrazole derivatives as potent JAK 2/3 and aurora A/B kinases multi-targeted inhibitors. Eur. J. Med. Chem 2021, 209, 112934.
- Ali, E.M.H.; El-Telbany, R.F.A.; Abdel-Maksoud, M.S.; Ammar, U.M.; Mersal, K.I.; Zaraei, S.O.; El-Gamal, M.I.; Choi, S.I.; Lee, K.T.; Kim, H.K.; et al. Design, synthesis, biological evaluation, and docking studies of novel (imidazol-5-yl)pyrimidine-based derivatives as dual BRAFV600E/p38α inhibitors. Eur. J. Med. Chem. 2021, 215, 113277.
- Lei, Y.; Zhang, B.; Zhang, Y.; Dai, X.; Duan, Y.; Mao, Q.; Gao, J.; Yang, Y.; Bao, Z.; Fu, X.; et al. Design, synthesis and biological evaluation of novel FXIa inhibitors with 2-phenyl-1H-imidazole-5-carboxamide moiety as P1 fragment. Eur. J. Med. Chem. 2021, 220, 113437.
- Sekioka, R.; Honda, S.; Akashiba, H.; Yarimizu, J.; Mitani, Y.; Yamasaki, S. Optimization and biological evaluation of imidazopyridine derivatives as a novel scaffold for γ-secretase modulators with oral efficacy against cognitive deficits in Alzheimer’s disease model mice. Bioorg. Med. Chem. 2020, 28, 115455.
- Yang, Y.; Chen, P.; Zhao, L.; Zhang, B.; Xu, C.; Zhang, H.; Zhou, J. Design, synthesis and biological evaluation of imidazolopyridone derivatives as novel BRD4 inhibitors. Bioorg. Med. Chem. 2021, 29, 115857.
- Bu, H.; Yuan, X.; Wu, H.; Zhou, J.; Zhang, H. Design, synthesis and biological evaluation of imidazopyridazine derivatives containing isoquinoline group as potent MNK1/2 inhibitors. Bioorg. Med. Chem. 2021, 40, 116186.
More
Information
Subjects:
Chemistry, Medicinal; Chemistry, Organic
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
5.0K
Entry Collection:
Organic Synthesis
Revisions:
4 times
(View History)
Update Date:
16 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No