 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dana Jurkovicova | -- | 4872 | 2022-08-10 10:29:50 | | | |
| 2 | Peter Tang | Meta information modification | 4872 | 2022-08-10 10:43:24 | | |
Video Upload Options
Mitochondria are dynamic organelles managing crucial processes of cellular metabolism and bioenergetics. Enabling rapid cellular adaptation to altered endogenous and exogenous environments, mitochondria play an important role in many pathophysiological states, including cancer. Being under the control of mitochondrial and nuclear DNA (mtDNA and nDNA), mitochondria adjust their activity and biogenesis to cell demands. In cancer, numerous mutations in mtDNA have been detected, which do not inactivate mitochondrial functions but rather alter energy metabolism to support cancer cell growth. Increasing evidence suggests that mtDNA mutations, mtDNA epigenetics and miRNA regulations dynamically modify signalling pathways in an altered microenvironment, resulting in cancer initiation and progression and aberrant therapy response.
1. Introduction
2. Mitochondrial Genetics and Cancer
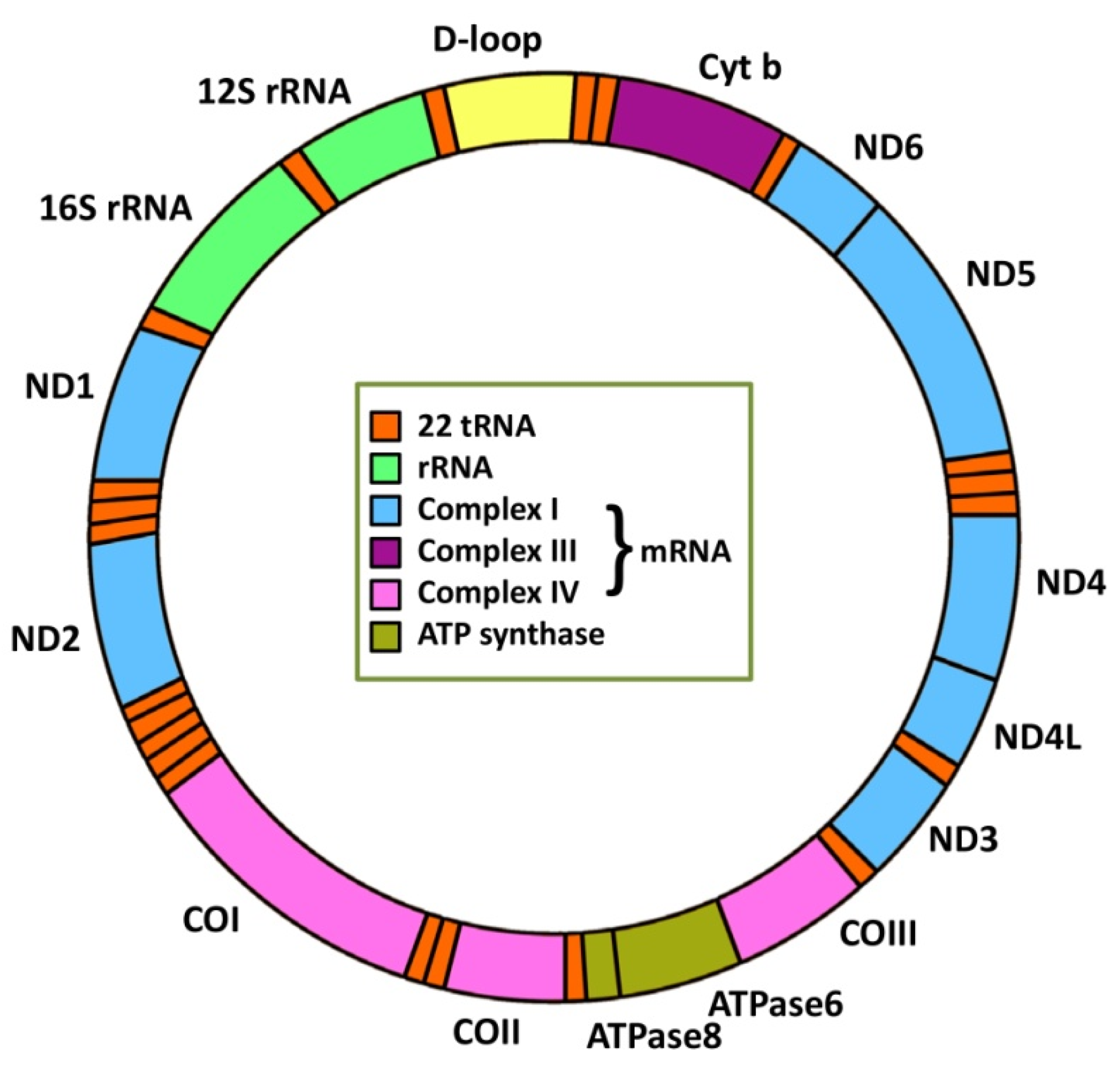
3. Epigenetic Regulation of Mitochondrial Genes in Cancer
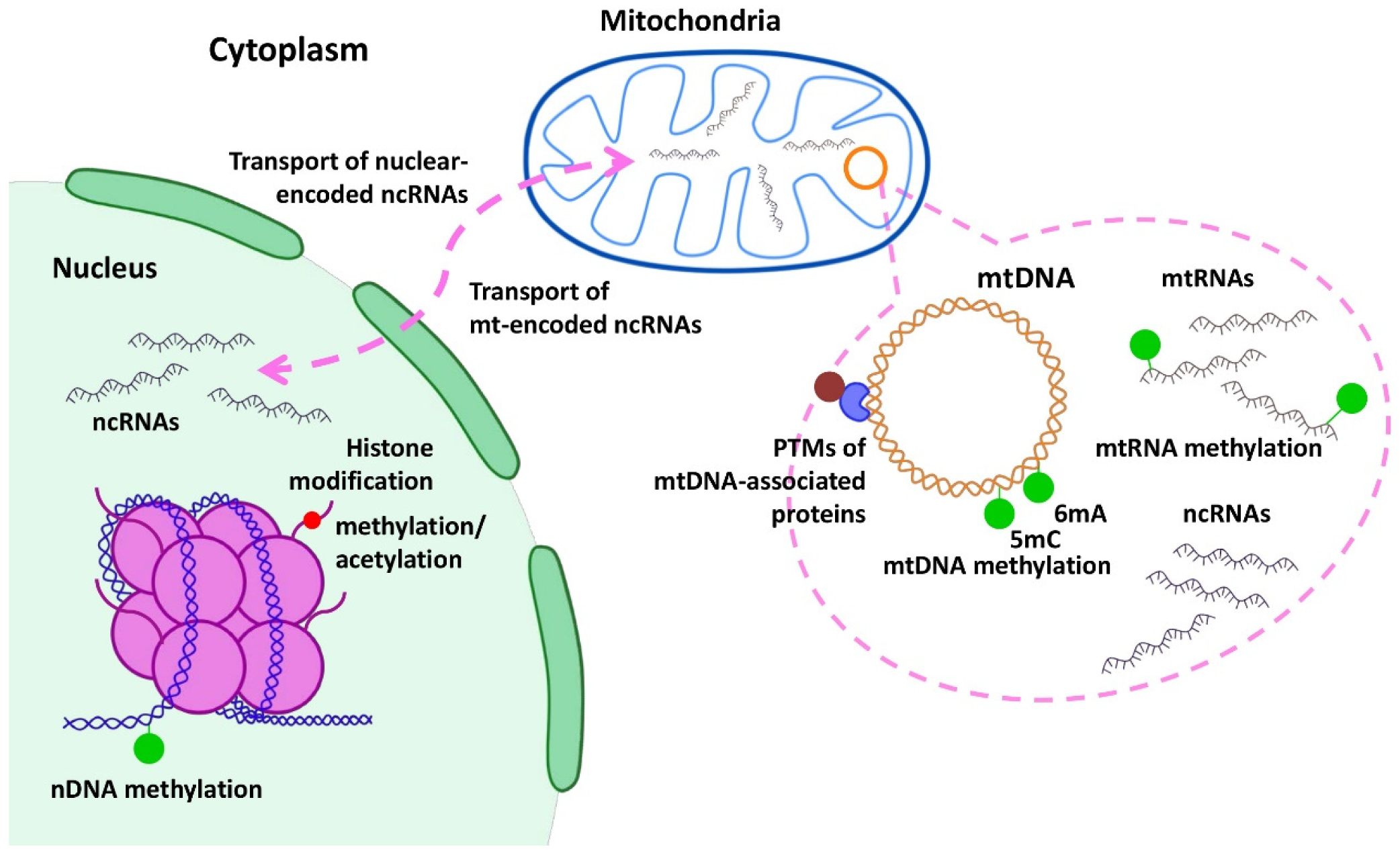
4. MiRNA Regulation of Mitochondrial Genes
5. Targeting Mitochondria to Combat Cancer
6. Therapeutic Potential
References
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036.
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467.
- Wallace, D.C. Why Do We Still Have a Maternally Inherited Mitochondrial DNA? Insights from Evolutionary Medicine. Annu. Rev. Biochem. 2007, 76, 781–821.
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102.
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754.
- Ma, Y.; Wang, L.; Jia, R. The role of mitochondrial dynamics in human cancers. Am. J. Cancer Res. 2020, 10, 1278–1293.
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314.
- Cassim, S.; Vučetić, M.; Ždralević, M.; Pouyssegur, J. Warburg and Beyond: The Power of Mitochondrial Metabolism to Collaborate or Replace Fermentative Glycolysis in Cancer. Cancers 2020, 12, 1119.
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566.
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157.
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308.
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic Signatures Uncover Distinct Targets in Molecular Subsets of Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 22, 547–560.
- Goto, M.; Miwa, H.; Shikami, M.; Tsunekawa-Imai, N.; Suganuma, K.; Mizuno, S.; Takahashi, M.; Mizutani, M.; Hanamura, I.; Nitta, M. Importance of Glutamine Metabolism in Leukemia Cells by Energy Production through TCA Cycle and by Redox Homeostasis. Cancer Investig. 2014, 32, 241–247.
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315.
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551.
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563.
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473.
- Cannino, G.; Ciscato, F.; Masgras, I.; Martin, C.S.; Rasola, A. Metabolic Plasticity of Tumor Cell Mitochondria. Front. Oncol. 2018, 8, 333.
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2020, 595, 976–1002.
- Gray, M.W. Mitochondrial Evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403.
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465.
- Cotter, D. MitoProteome: Mitochondrial protein sequence database and annotation system. Nucleic Acids Res. 2004, 32, D463–D467.
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286.
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- Lopes, A.F.C. Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin. Epigenet. 2020, 12, 182.
- Bellizzi, D.; D’Aquila, P.; Giordano, M.; Montesanto, A.; Passarino, G. Global DNA methylation levels are modulated by mitochondrial DNA variants. Epigenomics 2012, 4, 17–27.
- Manev, H.; Dzitoyeva, S. Progress in mitochondrial epigenetics. Biomol. Concepts 2013, 4, 381–389.
- Cyr, A.R.; Domann, F.E. The Redox Basis of Epigenetic Modifications: From Mechanisms to Functional Consequences. Antioxid. Redox Signal. 2011, 15, 551–589.
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31.
- Bohovych, I.; Khalimonchuk, O. Sending Out an SOS: Mitochondria as a Signaling Hub. Front. Cell Dev. Biol. 2016, 4, 109.
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226.
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34.
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655.
- Leung, A.K.L. The Whereabouts of miRNA Actions: Cytoplasm and Beyond miRNA: A Moving Target HHS Public Access. Trends Cell Biol. 2015, 25, 601–610.
- Bandiera, S.; Rüberg, S.; Girard, M.; Cagnard, N.; Hanein, S.; Chrétien, D.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. Nuclear Outsourcing of RNA Interference Components to Human Mitochondria. PLoS ONE 2011, 6, e20746.
- Bandiera, S.; Matégot, R.; Girard, M.; Demongeot, J.; Henrion-Caude, A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic. Biol. Med. 2013, 64, 12–19.
- Wallace, L.; Aikhionbare, K.; Banerjee, S.; Peagler, K.; Pitts, M.; Yao, X.; Aikhionbare, F. Differential Expression Profiles of Mitogenome Associated MicroRNAs Among Colorectal Adenomatous Polyps. Cancer Res. J. 2021, 9, 23.
- Lin, H.-Y.; Chu, P.-Y. Advances in Understanding Mitochondrial MicroRNAs (mitomiRs) on the Pathogenesis of Triple-Negative Breast Cancer (TNBC). Oxidative Med. Cell. Longev. 2021, 2021, 5517777.
- Qu, C.; Yan, C.; Cao, W.; Li, F.; Qu, Y.; Guan, K.; Si, C.; Yu, Z.; Qu, Z. miR-128-3p contributes to mitochondrial dysfunction and induces apoptosis in glioma cells via targeting pyruvate dehydrogenase kinase 1. IUBMB Life 2020, 72, 465–475.
- Fan, L.; Zhu, C.; Qiu, R.; Zan, P.; Zheng, Z.; Xu, T.; Li, G. MicroRNA-661 Enhances TRAIL or STS Induced Osteosarcoma Cell Apoptosis by Modulating the Expression of Cytochrome c1. Cell. Physiol. Biochem. 2017, 41, 1935–1946.
- He, Z.; Li, Z.; Zhang, X.; Yin, K.; Wang, W.; Xu, Z.; Li, B.; Zhang, L.; Xu, J.; Sun, G.; et al. MiR-422a regulates cellular metabolism and malignancy by targeting pyruvate dehydrogenase kinase 2 in gastric cancer. Cell Death Dis. 2018, 9, 505.
- Herr, I.; Sähr, H.; Zhao, Z.; Yin, L.; Omlor, G.; Lehner, B.; Fellenberg, J. MiR-127 and miR-376a act as tumor suppressors by in vivo targeting of COA1 and PDIA6 in giant cell tumor of bone. Cancer Lett. 2017, 409, 49–55.
- Fellenberg, J.; Lehner, B.; Saehr, H.; Schenker, A.; Kunz, P. Tumor Suppressor Function of miR-127-3p and miR-376a-3p in Osteosarcoma Cells. Cancers 2019, 11, 2019.
- Zhao, X.; Wang, Q.; Lin, F.; Wang, X.; Wang, Y.; Wang, J.; Wang, C. RNA Sequencing of Osteosarcoma Gene Expression Profile Revealed that miR-214-3p Facilitates Osteosarcoma Cell Proliferation via Targeting Ubiquinol-Cytochrome c Reductase Core Protein 1 (UQCRC1). Med Sci. Monit. 2019, 25, 4982–4991.
- Wang, D.-W.; Su, F.; Zhang, T.; Yang, T.-C.; Wang, H.-Q.; Yang, L.-J.; Zhou, F.-F.; Feng, M.-H. The miR-370/UQCRC2 axis facilitates tumorigenesis by regulating epithelial-mesenchymal transition in Gastric Cancer. J. Cancer 2020, 11, 5042–5055.
- Jung, K.-A.; Lee, S.; Kwak, M.-K. NFE2L2/NRF2 Activity Is Linked to Mitochondria and AMP-Activated Protein Kinase Signaling in Cancers Through miR-181c/Mitochondria-Encoded Cytochrome c Oxidase Regulation. Antioxidants Redox Signal. 2017, 27, 945–961.
- Zhang, J.; Liang, J.; Huang, J. Downregulated microRNA-26a modulates prostate cancer cell proliferation and apoptosis by targeting COX-2. Oncol. Lett. 2016, 12, 3397–3402.
- Zhuang, X.; Chen, Y.; Wu, Z.; Xu, Q.; Chen, M.; Shao, M.; Cao, X.; Zhou, Y.; Xie, M.; Shi, Y.; et al. Mitochondrial miR-181a-5p promotes glucose metabolism reprogramming in liver cancer by regulating the electron transport chain. Carcinogenesis 2019, 41, 972–983.
- Chen, W.; Wang, P.; Lu, Y.; Jin, T.; Lei, X.; Liu, M.; Zhuang, P.; Liao, J.; Lin, Z.; Li, B.; et al. Decreased expression of mitochondrial miR-5787 contributes to chemoresistance by reprogramming glucose metabolism and inhibiting MT-CO3 translation. Theranostics 2019, 9, 5739–5754.
- Owa, C.; Poulin, M.; Yan, L.; Shioda, T. Technical adequacy of bisulfite sequencing and pyrosequencing for detection of mitochondrial DNA methylation: Sources and avoidance of false-positive detection. PLoS ONE 2018, 13, e0192722.
- Purohit, P.K.; Edwards, R.; Tokatlidis, K.; Saini, N. MiR-195 regulates mitochondrial function by targeting mitofusin-2 in breast cancer cells. RNA Biol. 2019, 16, 918–929.
- Pan, L.; Zhou, L.; Yin, W.; Bai, J.; Liu, R. miR-125a induces apoptosis, metabolism disorder and migrationimpairment in pancreatic cancer cells by targeting Mfn2-related mitochondrial fission. Int. J. Oncol. 2018, 53, 124–136.
- Zhou, X.; Zhang, L.; Zheng, B.; Yan, Y.; Zhang, Y.; Xie, H.; Zhou, L.; Zheng, S.; Wang, W. Micro RNA-761 is upregulated in hepatocellular carcinoma and regulates tumorigenesis by targeting Mitofusin-2. Cancer Sci. 2016, 107, 424–432.
- Luan, T.; Fu, S.; Huang, L.; Zuo, Y.; Ding, M.; Li, N.; Chen, J.; Wang, H.; Wang, J. MicroRNA-98 promotes drug resistance and regulates mitochondrial dynamics by targeting LASS2 in bladder cancer cells. Exp. Cell Res. 2018, 373, 188–197.
- Li, B.; Wang, W.; Li, Z.; Chen, Z.; Zhi, X.; Xu, J.; Li, Q.; Wang, L.; Huang, X.; Wang, L.; et al. MicroRNA-148a-3p enhances cisplatin cytotoxicity in gastric cancer through mitochondrial fission induction and cyto-protective autophagy suppression. Cancer Lett. 2017, 410, 212–227.
- Fan, S.; Chen, W.-X.; Lv, X.-B.; Tang, Q.-L.; Sun, L.-J.; Liu, B.-D.; Zhong, J.-L.; Lin, Z.-Y.; Wang, Y.-Y.; Li, Q.-X.; et al. miR-483-5p determines mitochondrial fission and cisplatin sensitivity in tongue squamous cell carcinoma by targeting FIS1. Cancer Lett. 2015, 362, 183–191.
- Fan, S.; Liu, B.; Sun, L.; Lv, X.-B.; Lin, Z.; Chen, W.; Chen, W.; Tang, Q.; Wang, Y.; Su, Y.; et al. Mitochondrial fission determines cisplatin sensitivity in tongue squamous cell carcinoma through the BRCA1-miR-593-5p–MFF axis. Oncotarget 2015, 6, 14885–14904.
- Strappazzon, F. A global view of the miRNA-mitophagy connexion. In Progress in Molecular Biology and Translational Science, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; pp. 37–54.
- Di Rita, A.; Maiorino, T.; Bruqi, K.; Volpicelli, F.; Bellenchi, G.C.; Strappazzon, F. miR-218 Inhibits Mitochondrial Clearance by Targeting PRKN E3 Ubiquitin Ligase. Int. J. Mol. Sci. 2020, 21, 355.
- Tai, Y.; Pu, M.; Yuan, L.; Guo, H.; Qiao, J.; Lu, H.; Wang, G.; Chen, J.; Qi, X.; Tao, Z.; et al. miR-34a-5p regulates PINK1-mediated mitophagy via multiple modes. Life Sci. 2021, 276, 119415.
- Zhou, J.; Zhao, Y.; Li, Z.; Zhu, M.; Wang, Z.; Li, Y.; Xu, T.; Feng, D.; Zhang, S.; Tang, F.; et al. miR-103a-3p regulates mitophagy in Parkinson’s disease through Parkin/Ambra1 signaling. Pharmacol. Res. 2020, 160, 105197.
- Tsujimoto, T.; Mori, T.; Houri, K.; Onodera, Y.; Takehara, T.; Shigi, K.; Nakao, S.; Teramura, T.; Fukuda, K. miR-155 inhibits mitophagy through suppression of BAG5, a partner protein of PINK1. Biochem. Biophys. Res. Commun. 2020, 523, 707–712.
- Cheng, M.; Liu, L.; Lao, Y.; Liao, W.; Liao, M.; Luo, X.; Wu, J.; Xie, W.; Zhang, Y.; Xu, N. MicroRNA-181a suppresses parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis. Oncotarget 2016, 7, 42274–42287.
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.-G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 Is a Novel Hypoxia-responsive MicroRNA That Inhibits Mitophagy via Regulation of Two Mitophagy Receptors FUNDC1 and NIX. J. Biol. Chem. 2014, 289, 10691–10701.
- Liu, H.; Huang, H.; Li, R.; Bi, W.; Feng, L.; Lingling, E.; Hu, M.; Wen, W. Mitophagy protects SH-SY5Y neuroblastoma cells against the TNFα-induced inflammatory injury: Involvement of microRNA-145 and Bnip3. Biomed. Pharmacother. 2019, 109, 957–968.
- Singh, R.; Saini, N. Downregulation of BCL2 by miRNAs augments drug induced apoptosis: Combined computational and experimental approach. J. Cell Sci. 2012, 125, 1568–1578.
- Slattery, M.L.; Mullany, L.E.; Sakoda, L.C.; Wolff, R.K.; Samowitz, W.S.; Herrick, J.S. Dysregulated genes and miRNAs in the apoptosis pathway in colorectal cancer patients. Apoptosis 2018, 23, 237–250.
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949.
- Xie, Q.; Wang, S.; Zhao, Y.; Zhang, Z.; Qin, C.; Yang, X. MiR-519d impedes cisplatin-resistance in breast cancer stem cells by down-regulating the expression of MCL-1. Oncotarget 2017, 8, 22003–22013.
- Dang, K.; Myers, K.A. The Role of Hypoxia-Induced miR-210 in Cancer Progression. Int. J. Mol. Sci. 2015, 16, 6353–6372.
- Fuhrmann, D.C.; Brüne, B. Mitochondrial Composition and Function Under the Control of Hypoxia. Redox Biol. 2017, 12, 208–215.
- Nallamshetty, S.; Chan, S.Y.; Loscalzo, J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 2013, 64, 20–30.
- Koehler, J.; Sandey, M.; Prasad, N.; Levy, S.A.; Wang, X.; Wang, X. Differential Expression of miRNAs in Hypoxia (“HypoxamiRs”) in Three Canine High-Grade Glioma Cell Lines. Front. Veter-Sci. 2020, 7, 104.
- Zhang, W.; Chen, J.-H.; Shan, T.; Aguilera-Barrantes, I.; Wang, L.-S.; Huang, T.H.-M.; Rader, J.S.; Sheng, X.; Huang, Y.-W. miR-137 is a tumor suppressor in endometrial cancer and is repressed by DNA hypermethylation. Lab. Investig. 2018, 98, 1397–1407.
- Bi, W.; Xia, M.; Wang, X. miR-137 suppresses proliferation, migration and invasion of colon cancer cell lines by targeting TCF4. Oncol. Lett. 2018, 15, 8744–8748.
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472.
- Carden, T.; Singh, B.; Mooga, V.; Bajpai, P.; Singh, K.K. Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J. Biol. Chem. 2017, 292, 20694–20706.
- Macharia, L.W.; Wanjiru, C.M.; Mureithi, M.W.; Pereira, C.M.; Ferrer, V.P.; Moura-Neto, V. MicroRNAs, Hypoxia and the Stem-Like State as Contributors to Cancer Aggressiveness. Front. Genet. 2019, 10, 125.
- Wang, K.; Chen, Y.; Zhao, Z.; Feng, M.; Zhang, S. Identification of potential core genes and miRNAs in testicular seminoma via bioinformatics analysis. Mol. Med. Rep. 2019, 20, 4013–4022.
- Geiger, J.; Dalgaard, L.T. Interplay of mitochondrial metabolism and microRNAs. Cell. Mol. Life Sci. 2017, 74, 631–646.
- Duarte, F.V.; Palmeira, C.M.; Rolo, A.P. The Role of microRNAs in Mitochondria: Small Players Acting Wide. Genes 2014, 5, 865–886.
- Shinde, S.; Bhadra, U. A Complex Genome-MicroRNA Interplay in Human Mitochondria. BioMed Res. Int. 2015, 2015, 206382.
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614.
- Choudhury, A.R.; Singh, K.K. Mitochondrial determinants of cancer health disparities. Semin. Cancer Biol. 2017, 47, 125–146.
- Khwairakpam, A.D.; Shyamananda, M.S.; Sailo, B.L.; Rathnakaram, S.R.; Padmavathi, G.; Kotoky, J.; Kunnumakkara, A.B. ATP citrate lyase (ACLY): A promising target for cancer prevention and treatment. Curr. Drug Targets 2015, 16, 156–163.
- Wang, J.; Ye, W.; Yan, X.; Guo, Q.; Ma, Q.; Lin, F.; Huang, J.; Jin, J. Low expression of ACLY associates with favorable prognosis in acute myeloid leukemia. J. Transl. Med. 2019, 17, 149.
- Wang, Y.; Wang, Y.; Shen, L.; Pang, Y.; Qiao, Z.; Liu, P. Prognostic and therapeutic implications of increased ATP citrate lyase expression in human epithelial ovarian cancer. Oncol. Rep. 2012, 27, 1156–1162.
- Wei, X.; Shi, J.; Lin, Q.; Ma, X.; Pang, Y.; Mao, H.; Li, R.; Lu, W.; Wang, Y.; Liu, P. Targeting ACLY Attenuates Tumor Growth and Acquired Cisplatin Resistance in Ovarian Cancer by Inhibiting the PI3K–AKT Pathway and Activating the AMPK–ROS Pathway. Front. Oncol. 2021, 11, 642229.
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate Dependence of Tumors. Cell 2014, 159, 1591–1602.
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA Synthetase 2 Promotes Acetate Utilization and Maintains Cancer Cell Growth under Metabolic Stress. Cancer Cell 2015, 27, 57–71.
- Li, X.; Yu, W.; Qian, X.; Xia, Y.; Zheng, Y.; Lee, J.-H.; Li, W.; Lyu, J.; Rao, G.; Zhang, X.; et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol. Cell 2017, 66, 684–697.e9.
- Kargbo, R.B. Inhibition of ACSS2 for Treatment of Cancer and Neuropsychiatric Diseases. ACS Med. Chem. Lett. 2019, 10, 1100–1101.
- Lakhter, A.J.; Hamilton, J.; Konger, R.L.; Brustovetsky, N.; Broxmeyer, H.E.; Naidu, S.R. Glucose-independent Acetate Metabolism Promotes Melanoma Cell Survival and Tumor Growth. J. Biol. Chem. 2016, 291, 21869–21879.
- Vendramin, R.; Marine, J.-C.; Leucci, E. Non-coding RNA s: The dark side of nuclear–mitochondrial communication. EMBO J. 2017, 36, 1123–1133.
- Liu, H.; Lei, C.; He, Q.; Pan, Z.; Xiao, D.; Tao, Y. Nuclear functions of mammalian MicroRNAs in gene regulation, immunity and cancer. Mol. Cancer 2018, 17, 64.
- Zhang, X.; Zuo, X.; Yang, B.; Li, Z.; Xue, Y.; Zhou, Y.; Huang, J.; Zhao, X.; Zhou, J.; Yan, Y.; et al. MicroRNA Directly Enhances Mitochondrial Translation during Muscle Differentiation. Cell 2014, 158, 607–619.
- Das, S.; Bedja, D.; Campbell, N.; Dunkerly, B.; Chenna, V.; Maitra, A.; Steenbergen, C. miR-181c Regulates the Mitochondrial Genome, Bioenergetics, and Propensity for Heart Failure In Vivo. PLoS ONE 2014, 9, e96820.
- Das, S.; Ferlito, M.; Kent, O.; Fox-Talbot, K.; Wang, R.; Liu, D.; Raghavachari, N.; Yang, Y.; Wheelan, S.J.; Murphy, E.; et al. Nuclear miRNA Regulates the Mitochondrial Genome in the Heart. Circ. Res. 2012, 110, 1596–1603.
- Ji, W.; Sun, B.; Su, C. Targeting MicroRNAs in Cancer Gene Therapy. Genes 2017, 8, 21.
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704.
- Shah, V.; Shah, J. Recent trends in targeting miRNAs for cancer therapy. J. Pharm. Pharmacol. 2020, 72, 1732–1749.
- Gokita, K.; Inoue, J.; Ishihara, H.; Kojima, K.; Inazawa, J. Therapeutic Potential of LNP-Mediated Delivery of miR-634 for Cancer Therapy. Mol. Ther.-Nucleic Acids 2020, 19, 330–338.
- O’Brien, K.P.; Khan, S.; Gilligan, K.; Zafar, H.; Lalor, P.; Glynn, C.; O’Flatharta, C.; Ingoldsby, H.; Dockery, P.; De Bhulbh, A.; et al. Employing mesenchymal stem cells to support tumor-targeted delivery of extracellular vesicle (EV)-encapsulated microRNA-379. Oncogene 2018, 37, 2137–2149.
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204.
- Yan, L.X.; Wu, Q.N.; Zhang, Y.; Li, Y.Y.; Liao, D.Z.; Hou, J.H.; Fu, J.; Zeng, M.S.; Yun, J.P.; Wu, Q.L.; et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivotumor growth. Breast Cancer Res. 2011, 13, R2.
- Yin, H.; Xiong, G.; Guo, S.; Xu, C.; Xu, R.; Guo, P.; Shu, D. Delivery of Anti-miRNA for Triple-Negative Breast Cancer Therapy Using RNA Nanoparticles Targeting Stem Cell Marker CD133. Mol. Ther. 2019, 27, 1252–1261.
- Kunz, M.; Brandl, M.; Bhattacharya, A.; Nobereit-Siegel, L.; Ewe, A.; Weirauch, U.; Hering, D.; Reinert, A.; Kalwa, H.; Guzman, J.; et al. Nanoparticle-complexed antimiRs for inhibiting tumor growth and metastasis in prostate carcinoma and melanoma. J. Nano. 2020, 18, 173.
- Yang, H.; Liu, Y.; Qiu, Y.; Ding, M.; Zhang, Y. MiRNA-204-5p and oxaliplatin-loaded silica nanoparticles for enhanced tumor suppression effect in CD44-overexpressed colon adenocarcinoma. Int. J. Pharm. 2019, 566, 585–593.
- Sukumar, U.K.; Bose, R.J.C.; Malhotra, M.; Babikir, H.A.; Afjei, R.; Robinson, E.; Zeng, Y.; Chang, E.; Habte, F.; Sinclair, R.; et al. Intranasal delivery of targeted polyfunctional gold–iron oxide nanoparticles loaded with therapeutic microRNAs for combined theranostic multimodality imaging and presensitization of glioblastoma to temozolomide. Biomaterials 2019, 218, 119342.
- Rachek, L.I.; Grishko, V.I.; Musiyenko, S.I.; Kelley, M.R.; LeDoux, S.P.; Wilson, G.L. Conditional Targeting of the DNA Repair Enzyme hOGG1 into Mitochondria. J. Biol. Chem. 2002, 277, 44932–44937.
- Rachek, L.I.; Grishko, V.I.; Alexeyev, M.F.; Pastukh, V.V.; LeDoux, S.P.; Wilson, G.L. Endonuclease III and endonuclease VIII conditionally targeted into mitochondria enhance mitochondrial DNA repair and cell survival following oxidative stress. Nucleic Acids Res. 2004, 32, 3240–3247.
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; Seidman, J.G.; et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091.
- Neuzil, J.; Dong, L.-F.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208.
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120.
- Modica-Napolitano, J.S.; Aprille, J.R. Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev. 2001, 49, 63–70.
- Houston, M.A.; Augenlicht, L.H.; Heerdt, B.G. Stable Differences in Intrinsic Mitochondrial Membrane Potential of Tumor Cell Subpopulations Reflect Phenotypic Heterogeneity. Int. J. Cell Biol. 2011, 2011, 978583.
- Murphy, M.P. Targeting lipophilic cations to mitochondria. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1028–1031.
- Smith, R.A.J.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412.
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Sikora, A.; Zielonka, J.; Dwinell, M. Mitochondria-targeted metformins: Anti-tumour and redox signalling mechanisms. Interface Focus 2017, 7, 20160109.
- Millard, M.; Gallagher, J.D.; Olenyuk, B.Z.; Neamati, N. A Selective Mitochondrial-Targeted Chlorambucil with Remarkable Cytotoxicity in Breast and Pancreatic Cancers. J. Med. Chem. 2013, 56, 9170–9179.
- Han, M.; Vakili, M.R.; Abyaneh, H.S.; Molavi, O.; Lai, R.; Lavasanifar, A. Mitochondrial Delivery of Doxorubicin via Triphenylphosphine Modification for Overcoming Drug Resistance in MDA-MB-435/DOX Cells. Mol. Pharm. 2014, 11, 2640–2649.
- Spivak, A.Y.; Nedopekina, D.A.; Gubaidullin, R.R.; Davletshin, E.V.; Tukhbatullin, A.A.; D’Yakonov, V.A.; Yunusbaeva, M.M.; Dzhemileva, L.U.; Dzhemilev, U.M. Pentacyclic triterpene acid conjugated with mitochondria-targeting cation F16: Synthesis and evaluation of cytotoxic activities. Med. Chem. Res. 2021, 30, 940–951.
- Spivak, A.Y.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Dzhemilev, U.M.; Bel’skii, Y.P.; Bel’skaya, N.V.; Stankevich, S.A.; et al. Synthesis of lupane triterpenoids with triphenylphosphonium substituents and studies of their antitumor activity. Russ. Chem. Bull. 2013, 62, 188–198.
- Nedopekina, D.A.; Gubaidullin, R.R.; Odinokov, V.N.; Maximchik, P.V.; Zhivotovsky, B.; Bel’Skii, Y.P.; Khazanov, V.A.; Manuylova, A.V.; Gogvadze, V.; Spivak, A.Y. Mitochondria-targeted betulinic and ursolic acid derivatives: Synthesis and anticancer activity. MedChemComm 2017, 8, 1934–1945.
- Spivak, A.Y.; Nedopekina, D.A.; Khalitova, R.R.; Gubaidullin, R.R.; Odinokov, V.N.; Bel’Skii, Y.P.; Bel’Skaya, N.V.; Khazanov, V.A. Triphenylphosphonium cations of betulinic acid derivatives: Synthesis and antitumor activity. Med. Chem. Res. 2017, 26, 518–531.
- Tsepaeva, O.V.; Nemtarev, A.V.; Abdullin, T.I.; Grigor’Eva, L.R.; Kuznetsova, E.V.; Akhmadishina, R.A.; Ziganshina, L.E.; Cong, H.H.; Mironov, V.F. Design, Synthesis, and Cancer Cell Growth Inhibitory Activity of Triphenylphosphonium Derivatives of the Triterpenoid Betulin. J. Nat. Prod. 2017, 80, 2232–2239.
- Ye, Y.; Zhang, T.; Yuan, H.; Li, D.; Lou, H.; Fan, P. Mitochondria-Targeted Lupane Triterpenoid Derivatives and Their Selective Apoptosis-Inducing Anticancer Mechanisms. J. Med. Chem. 2017, 60, 6353–6363.
- Hoenke, S.; Serbian, I.; Deigner, H.-P.; Csuk, R. Mitocanic Di- and Triterpenoid Rhodamine B Conjugates. Molecules 2020, 25, 5443.
- Heise, N.V.; Major, D.; Hoenke, S.; Kozubek, M.; Serbian, I.; Csuk, R. Rhodamine 101 Conjugates of Triterpenoic Amides Are of Comparable Cytotoxicity as Their Rhodamine B Analogs. Molecules 2022, 27, 2220.
- Dubinin, M.; Semenova, A.; Nedopekina, D.; Davletshin, E.; Spivak, A.; Belosludtsev, K. Effect of F16-Betulin Conjugate on Mitochondrial Membranes and Its Role in Cell Death Initiation. Membranes 2021, 11, 352.
- Spivak, A.; Nedopekina, D.; Gubaidullin, R.; Dubinin, M.; Belosludtsev, K. Conjugation of Natural Triterpenic Acids with Delocalized Lipophilic Cations: Selective Targeting Cancer Cell Mitochondria. J. Pers. Med. 2021, 11, 470.
- Muhammad, N.; Tan, C.-P.; Muhammad, K.; Wang, J.; Sadia, N.; Pan, Z.-Y.; Ji, L.-N.; Mao, Z.-W. Mitochondria-targeting monofunctional platinum(ii)–lonidamine conjugates for cancer cell de-energization. Inorg. Chem. Front. 2020, 7, 4010–4019.
- Ouyang, C.; Chen, L.; Rees, T.W.; Chen, Y.; Liu, J.; Ji, L.; Long, J.; Chao, H. A mitochondria-targeting hetero-binuclear Ir(iii)–Pt(ii) complex induces necrosis in cisplatin-resistant tumor cells. Chem. Commun. 2018, 54, 6268–6271.
- Zhang, C.; Guan, R.; Liao, X.; Ouyang, C.; Liu, J.; Ji, L.; Chao, H. Mitochondrial DNA targeting and impairment by a dinuclear Ir–Pt complex that overcomes cisplatin resistance. Inorg. Chem. Front. 2020, 7, 1864–1871.
- Marrache, S.; Pathak, R.K.; Dhar, S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 10444–10449.
- Wisnovsky, S.P.; Wilson, J.J.; Radford, R.J.; Pereira, M.P.; Chan, M.R.; Laposa, R.R.; Lippard, S.J.; Kelley, S.O. Targeting Mitochondrial DNA with a Platinum-Based Anticancer Agent. Chem. Biol. 2013, 20, 1323–1328.
- Dhar, S.; Daniel, W.L.; Giljohann, D.A.; Mirkin, C.A.; Lippard, S.J. Polyvalent Oligonucleotide Gold Nanoparticle Conjugates as Delivery Vehicles for Platinum(IV) Warheads. J. Am. Chem. Soc. 2009, 131, 14652–14653.
- Subhan, A.; Torchilin, V. siRNA based drug design, quality, delivery and clinical translation. Nanomed. Nano. Biol. Med. 2020, 29, 102239.
- Mun, J.-Y.; Baek, S.-W.; Park, W.Y.; Kim, W.-T.; Kim, S.-K.; Roh, Y.-G.; Jeong, M.-S.; Yang, G.-E.; Lee, J.-H.; Chung, J.W.; et al. E2F1 Promotes Progression of Bladder Cancer by Modulating RAD54L Involved in Homologous Recombination Repair. Int. J. Mol. Sci. 2020, 21, 9025.
- Mallick, A.; More, P.; Ghosh, S.; Chippalkatti, R.; Chopade, B.A.; Lahiri, M.; Basu, S. Dual Drug Conjugated Nanoparticle for Simultaneous Targeting of Mitochondria and Nucleus in Cancer Cells. ACS Appl. Mater. Interfaces 2015, 7, 7584–7598.
- Mallick, A.; More, P.; Syed, M.M.K.; Basu, S. Nanoparticle-Mediated Mitochondrial Damage Induces Apoptosis in Cancer. ACS Appl. Mater. Interfaces 2016, 8, 13218–13231.
- Akrami, M.; Samimi, S.; Alipour, M.; Bardania, H.; Ramezanpour, S.; Najafi, N.; Hosseinkhani, S.; Kamankesh, M.; Haririan, I.; Hassanshahi, F. Potential anticancer activity of a new pro-apoptotic peptide–thioctic acid gold nanoparticle platform. Nanotechnology 2021, 32, 145101.

