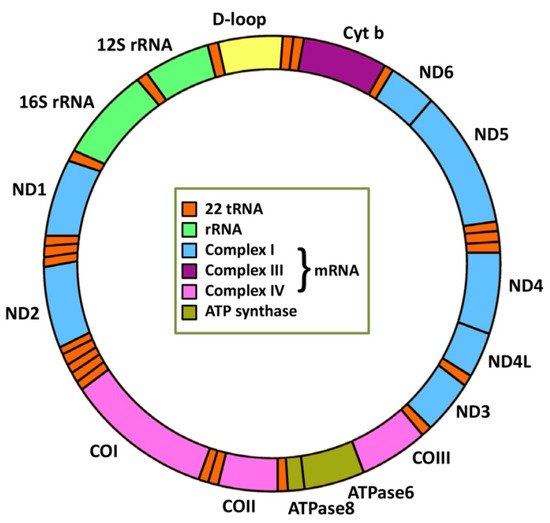
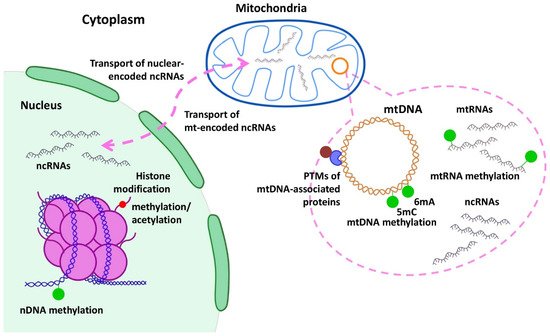
4. MiRNA Regulation of Mitochondrial Genes
MicroRNAs (miRNAs or miRs) represent a class of small (~22 nucleotides) non-coding RNAs transcribed from DNA sequences. They posttranscriptionally regulate gene expression and thus affect various cellular pathways, including those in mitochondria [
228,
229]. Modulation of mitochondrial processes by miRNA binding to mRNA occurs in the cytoplasm, or miRNAs are translocated into mitochondria, where they can directly target mitochondrial mRNAs. Moreover, human mitochondria themselves have been shown to possess miRNA sequences in their DNA, although their transcription from the mitochondrial genome remains to be fully clarified. MiRNA localizing in the mitochondria are referred to as “mitomiRs” and their activity mostly involves regulation of OXPHOS and ROS generation [
229,
230,
231].
In cancer, mitomiRs were shown to directly or indirectly regulate mitochondrial processes. For example, colorectal cancer expression profiles of mitogenome-associated miRNAs showed increased levels of miR-24, miR-181, miR-210, miR-21 and miR-378 [
232]. The oncogenic effects of miR-210, miR-155, miR-21, miR-224 and miR-373, and the anti-tumour action of miR-29a, miR-128, miR-342, miR-30a, miR-340, miR-18a and miR-224, have been associated with breast cancer pathologies (reviewed in [
233]). Microarray studies found significant changes in the expression of miR-7, miR-153, miR-21, miR-34a and miR-128 in malignant gliomas [
234].
Regarding metabolic reprogramming and OXPHOS regulation, miR-661 was shown to directly target cytochrome c1 and was downregulated in osteosarcoma cells [
235]. Furthermore, the glycolytic phenotype of glioma cells can be disrupted by miR-128-3p overexpression, which is able to modulate pyruvate dehydrogenase kinase 1 [
234]. On the other hand, pyruvate dehydrogenase kinase 2 was suggested to be a target for miR-422a when overexpressed in gastric cancer [
236]. The tumorigenicity of giant cell tumours of bone was demonstrated to be significantly reduced by restoration of miR-127-3p and miR-376a-3p expression, and this positive effect seems to be partially mediated via cytochrome c oxidase assembly factor 1 homolog (COA1) and protein disulphide isomerase family A member 6 (PDIA6) [
237]. A later study by the same group confirmed the role of miR-127 and miR-376a in tumour suppression in osteosarcoma cells [
238]. In contrast, osteosarcoma cell proliferation can be enhanced via targeting of ubiquinol-cytochrome c reductase core protein 1 (UQCRC1) with miR-214-3p [
239]. In gastric cancer, the miR-370/UQCRC2 axis positively regulates the epithelial-mesenchymal transition (EMT) signalling that affects tumour proliferation and metastasis [
240]. However, there is still limited information on direct regulation of mitochondria-encoded proteins by mitomiRs in cancer. Jung et al. [
241] confirmed that miR-181c, able to regulate the mitochondrial genome in rat cardiomyocytes, interacted with the mitochondrial gene encoding cytochrome c oxidase subunit 1 (MT-CO1). Reduction of miR-181c-mediated MT-CO1 was associated with mitochondrial membrane potential disruption, ATP reduction and adenosine monophosphate (AMP)-activated protein kinase-α (AMPKα) activation in human colon cancer cells [
241]. Expression of mitomiR miR-26a is decreased in prostate cancer compared to normal prostate cells, but its overexpression after transfection with miR-26a mimics significantly enhanced apoptosis via MT-CO2 inhibition [
242]. A recent report showed miR-181a-5p potentially targeting the
MT-CO2 and cytochrome b (
MT-CYB) genes in human hepatocellular carcinoma. Overexpression of miR-181a-5p reduced the level of MT-CYB and MT-CO2, impaired mitochondrial function and promoted glycolysis, leading to higher proliferation and metastasis [
243]. Differential expression microarray analysis identified downregulation of miR-5787 in cisplatin (CDDP)-resistant tongue squamous cell carcinoma (TSCC), resulting in suppression of MT-CO3 translation and a metabolic shift from OXPHOS to glycolysis [
244]. The CDDP-resistant phenotype of TSCC cells was found to be regulated by miR-2392. In chemoresistant patients, tumour expression of miR-2392 was increased and partially regulated transcription of mitochondrial genes in a splicing-competent Argonaute 2 (AGO2)-dependent manner [
139].
Several studies have indicated a connection between mitomiRs and mitochondrial dynamics.
MFN2 was shown to be a direct target of miR-195 in breast cancer cells. Mfn2 levels were significantly decreased when miR-195 was overexpressed and mitochondria became round, small and fragmented; however, miR-195-induced apoptosis was Mfn2-independent [
245]. In other study, negative regulation of
MFN2 by miR-125 led to induction of mitochondrial fission and promotion of pancreatic cancer cells’ apoptosis [
246]. Zhou et al. [
247], investigating the role of miR-761 in hepatocellular cancer cells in the context of mitochondrial dynamics, revealed that miR-761 overexpression directly downregulated Mfn2 and increased cell migration and invasion. When a miR-761 was inhibited, cancer cells were more receptive to apoptosis by upregulation of Mfn2 [
247]. In osteosarcoma cells, miR-19b was reported to directly target
MFN1 and suppress its anti-proliferative activity. On the other hand, inhibition of miR-19b promotes Mfn1-induced apoptosis. Several miRNAs have been shown to affect mitochondrial dynamics in chemoresistant cancer cells. MiR-98 and miR-148a-3p have been connected with regulation of Drp1 in resistant bladder and gastric cancers [
248,
249]. In TSCC, CDDP sensitivity was dependent on miR-483-5p regulation of Fis1 expression through a breast cancer type 1 susceptibility protein (BRCA1)–miR-593-5p–mitochondrial fission factor (Mff) axis [
250,
251].
MitomiRs, as far-reaching regulators, have been shown to also be directly linked to various mitophagy-associated proteins. Mitophagy represents a selective form of autophagy that cells use to remove dysfunctional or damaged mitochondria. In phosphatase and tensin homolog (PTEN)-induced kinase I/Parkin (PINK1/Parkin)-mediated mitophagy, miR-27a, miR-27b, miR-181a and miR-218 have been shown to regulate PINK1 and Parkin, but newer reports also indicate the involvement of miR-34a-5p, miR-103a-3p and miR-155 [
252,
253,
254,
255,
256]. In cancer, miR-181a suppresses Parkin-mediated mitophagy and sensitizes neuroblastoma cells to mitochondrial uncoupler-induced apoptosis [
257]. There is limited information on miRNA regulation of mitophagy receptors. MiR-137 has been reported to impair mitophagy in response to hypoxia via direct regulation of NIX (or BCL-2/adenovirus E1B 19 kDa-interacting protein 3 like (BNIP3L)) and Fun14 domain containing 1 (FUNDC1) receptors, and miR-145 has been suggested to be an upstream regulator of BNIP3-dependent mitophagy [
258,
259].
Several studies confirmed the ability of miRNAs to regulate proteins engaged in intrinsic apoptosis. In breast cancer, miR-195, miR-24-2 and miR-365-2 were shown to act as negative regulators of BCL-2 that enhance apoptosis [
260]. Slattery et al. associated miR-203a with BCL-2-mediated apoptosis in colorectal cancer [
261]. Chronic lymphocytic leukaemia exhibits deletion or downregulation of miR-15a and miR-16-1. Expression of these mitomiRs inhibits BCL-2-induced apoptosis [
262]. Overexpression of miR-519d enhances the sensitivity of CDDP-resistant breast cancer stem cells and mediates apoptosis through induced myeloid leukaemia cell differentiation protein (MCL-1)-dependent mitochondrial pathway [
263].
In hypoxia, HIF-modulated miR-210 is the major regulator of metabolism. MiR-210 reduces the ETC activities of complexes I and IV by disrupting electron flow through these chains and inducing the formation of ROS during hypoxia [
264,
265]. Overexpression of miR-210 was documented in pancreatic, head and neck, breast and lung cancers [
266]. Similar to miR-210, miR-323 is upregulated in hypoxia, and its low levels are connected to longer survival in human glioblastoma patients [
267]. Although miR-137 functions as a tumour suppressor [
268], it also plays a role in hypoxia and inhibition of ROS accumulation; miR-137 directly downregulates the expression of the hypoxia-mediated mitophagy receptors FUNDC1 and NIX, leading to suppression of apoptosis and mitophagy [
253]. Overexpression of miR-137 suppresses cell proliferation and migration [
269,
270]. MiR-216b inhibits proliferation and cell invasion in several types of cancer and is downregulated in hypoxia. Low levels of this miRNA were associated with poor prognosis [
267].
Mir-663 is necessary for stability of the respiratory complexes. It regulates expression of nuclear proteins involved in ETC in complexes I, II, III and IV. However, along with disruption of OXPHOS, expression of miR-663 is downregulated, showing mitochondria-to-nucleus retrograde signalling involving ROS. Increased miR-663 expression in patients with breast tumours correlated with increased survival [
271]. Overexpression of miR-128 reduced cell proliferation, angiogenesis and tumour growth through effects on HIF-1 and vascular endothelial growth factor (VEGF) [
272].
MiR-650, miR-665, miR-640, miR-1182, miR-1203, miR-661 and miR-1204 were all shown to be important in testicular cancer. MiR-650 and miR-665 were associated with the PI3K/AKT and Wnt/β-catenin signalling pathways, which participate in growth, migration and invasion of cancer cells. MiR-650 is downregulated in cancer and associated with inhibition of cell growth and invasion. This downregulation can activate AKT pathways and promote cell migration and proliferation. MiR-665 inhibits c-MYC and thus suppresses tumorigenesis. Tumour suppressor miR-665 is downregulated in cancer. Low levels of miR-661 and miR-640 are associated with poor cancer prognosis. MiR-640 affects the VEGF receptor 2-mTOR pathway. Upregulated miR-1204 promotes proliferation, glucose uptake and ATP production and correlates with tumour size [
273].
There is a significant number of miRNAs that affect mitochondrial processes [
274,
275,
276]. These mitomiRs, both oncogenic and tumour-suppressive, have been proposed as the key regulators of cancer-related processes, and their therapeutic targeting can be suggested as one of the key emerging novel diagnostic and therapeutic tools [
277].
5. Targeting Mitochondria to Combat Cancer
Given the massive involvement of mitochondria in cancer, therapeutic targeting of their genetic, epigenetic and miRNA landscape has a clear justification. Several molecular mechanisms of mitochondrial retrograde signalling have been revealed, such as those mediated by ROS, NAD
+/NADH and ATP, as well as those involving mitochondrial unfolding protein response, Ca
2+ gradient and calcineurin, AMP-activated protein kinase signalling and the “mipigenetic” process (mitochondrial–nuclear intergenomic crosstalk at the genetic and epigenetic level) [
57]. The retrograde signalling represents a causal factor in tumorigenesis but also induces invasive behaviour in tumour cells and contributes to tumour progression. Not surprisingly, targeting retrograde signalling mechanisms is of a great interest, as it provides another promising strategy for the development of selective cancer therapy.
In view of the regulation of the nucleo-cytoplasmic pool of acetyl-CoA, new promising therapeutic approaches have been envisioned. In this regard, the potential of
ACLY or
ACSS2 gene expression and activity modulation has been proposed. High levels of
ACLY expression associated with high proliferation rates have been detected in many types of tumours [
278], and this enzyme has been investigated as a prognostic factor for several cancers [
279,
280]. Wei et al. (2021) identified significantly upregulated ACLY in CDDP-resistant ovarian cancer cells and managed to sensitize the resistant cells by
ACLY knockdown. ACLY has been proposed as the key enzyme regulating acquired CDDP resistance in ovarian cancer and, therefore, as a suitable novel target for sensitization of ovarian tumours to platinum agents [
278,
281]. Another promising target for cancer therapy is ACSS2, the enzyme responsible for capturing acetate, the major source of acetyl-CoA, especially in hypoxia.
ACSS2 is highly expressed in numerous tumours and several studies have shown that genetic depletion or pharmacological targeting of ACSS2 inhibits tumour growth in breast, prostate, liver, pancreatic, ovarian and skin cancers and glioblastoma [
206,
207,
208,
282,
283].
MiRNAs modulate mitochondrial function via direct targeting of the mRNA of nuclear-encoded mitochondrial genes [
284]. Via their non-canonical functions, miRNAs may act as chromatin and transcriptional regulators in the nucleus (i.e., miR-584-3p and miR-26a-1) [
285] or as translational activators in mitochondria [
286]. In the mitochondrial genome, some mitomiRs’ seed sequences have been detected in silico, supporting regulation of mitochondrial transcripts encoded by mitomiRs [
287,
288]. In principle, miRNA-targeted therapies are based on either replenishing the tumour-suppressive miRNA mimetics, which enables restoration of the lost or downregulated tumour suppressor miRNA [
289], or administration of miRNA antagonists, antimiRs, targeting oncomiRs [
290,
291]. Accordingly, novel treatments based on targeting miRNAs have been proposed. For example, nanoparticles with miR-634 mimics caused a significant reduction in pancreatic tumour growth [
292]. Engineered extracellular vesicles released from the mesenchymal stem cells carrying miR-379 have been systematically administered to inhibit tumour growth in breast cancer [
293]. Similarly, marrow stromal cell exosomes carrying the miR-146b expression plasmid were successfully employed as an anti-tumour therapy in a rat model of glioblastoma [
294]. Nanoparticles carrying anti-miR-21 were able to selectively target triple-negative breast cancer (TNBC) cells to reduce miR-21 expression and activate apoptosis and proliferation control [
295,
296]. Inhibition of miR-150 and miR-638 was found to be efficient in reducing primary melanoma growth, as well as metastases [
297]. Treatment with combined oxaliplatin and miR-204-5p on silica nanoparticles significantly decreased growth of colon cancer [
298]. Anti-miR-21 and miR-100 on gold–iron oxide nanoparticles loaded with PEG-T7 peptide increased the overall survival rate in mice with glioblastoma multiforme when used in combination with systemic temozolomide administration [
299]. Today, there are numerous clinical trials investigating anti-miRNA sequences as miRNA-based cancer therapy, as monotherapy or as a combinatorial therapy. Targeting mitomiRs has huge therapeutic potential and, in parallel, mitomiRs represent a novel group of suitable diagnostic and prognostic markers.
With regard to the therapeutic focus on mtDNA mutations and their repair, it is evident that one of the promising, effective ways to prevent the onset and progression of human cancers associated with this organelle is elimination of the mtDNA mutations. As none of the DNA repair factors are encoded in mtDNA, efficiency in mtDNA repair can only be achieved by potential direction of more nuclear DNA repair factors to the mitochondria, although a few DNA repair factors possess mitochondrial targeting sequences. Indeed, targeting of DNA repair to mitochondria was shown to not only enhance the repair of mtDNA lesions but also increased the viability of treated cells and protected them against induction of apoptosis [
300,
301]. However, once an mtDNA mutation is fixed, its negative effect can be corrected only by introducing a construct carrying and expressing the wild-type version of the affected gene. Hence, one promising therapeutic approach for patients with mtDNA mutations is based on expression of the relevant proteins that are fused at the N-terminus with the mitochondrial targeting sequence. Unfortunately, no successful method has yet been introduced for clinically complementing mitochondrial dysfunction caused by mtDNA mutation in human mitochondrial disorders.
6. Therapeutic Potential
Over the last few years, mitochondria have proven to be an intriguing target for anti-cancer drugs. Indeed, due to their great clinical potential, anti-cancer agents targeting these organelles have become a highly prioritized focus of current cancer research. The exceptional potential of mitochondria to act as an anti-cancer target is strengthened by the facts that the abovementioned tumour-specific somatic mutations in mtDNA vary across tumour types [
302] and, importantly, that tumours of the same type but from different individuals may vary remarkably in the mutations functionally affecting mitochondria [
303]. Both facts point to an extraordinary possibility for the personalisation of anti-cancer therapy via targeting of mitochondria. Therefore, anti-cancer agents acting via mitochondrial destabilization, collectively referred to as mitocans (an acronym for “mitochondria and cancer”), represent one of the most innovative therapeutic approaches to drug targeting for the “next generation”. These agents have been divided into several classes based on the mode and site (from the surface of the outer mitochondrial membrane to the mitochondrial matrix) of action. Individual classes consist of the hexokinase inhibitors, compounds targeting the BCL-2 family proteins, thiol redox inhibitors, drugs targeting the voltage-dependent anionic channel and adenine nucleotide translocase, electron redox chain-targeting drugs, drugs targeting the TCA cycle, drugs targeting mtDNA and delocalized lipophilic cations (DLCs) targeting inner mitochondrial membrane [
303]. Selected examples of mitocans and their biological effects are described below, with particular focus on those covalently linking DLCs with widely used chemotherapeutics.
The highly negative plasma and mitochondrial membrane potentials (MMPs) (30–40 and 120–180 mV, respectively) permit 5–10 times higher concentrations of cations to be present in mitochondria than in the cytosol and 100–1000 times higher concentrations in the mitochondrial matrix than in the cytosol [
304,
305]. Based on this and on the fact that cancer cells have a more hyperpolarized MMP (app. 220 mV) [
306], several delocalized lipophilic cations (DLCs) have been introduced into experimental cancer research to improve mitochondrial uptake of anti-cancer drugs of interest [
304]. At the beginning, however, the use of DLCs in mitochondrial targeting was primarily developed to study mitochondrial physiology and dysfunction and the interaction between mitochondria and other subcellular organelles. Subsequently, many other applications for DLC-mediated mitochondrial targeting were revealed including those aimed at developing new therapy strategies in the field of cancer [
304,
307].
Triphenylphosphonium cation (TPP
+) is the most extensively used mitochondria targeting DLC structure and can easily pass through phospholipid bilayers because its charge is dispersed over a large surface area and the potential gradient drives its accumulation into mitochondrial matrix. When administered orally, it is able to pass from the gut to the bloodstream, within which it rapidly redistributes into organs. Direct intravenous injection can also be used to deliver the TPP
+-conjugated compounds to the mitochondria within cells in an organism. Importantly, orally administered TPP
+-conjugated compounds can be bioavailable, as shown for TPP
+ coupled to a coenzyme Q or vitamin E derivative in mice, where significant doses of these compounds could be fed safely over long periods, leading to steady-state distributions within several tissue/organ types. This indicates that therapeutic concentrations of the TPP
+-conjugated compounds can be reached by oral administration in tissues affected by mitochondrial dysfunction [
308], with the caveat that the toxic effects due to non-specific disruption to mitochondria caused by accumulation of large amounts of the DLCs may represent the major factor limiting the amount of the compounds that can be administered safely. Tethering TPP
+ to metformin significantly reduces human pancreatic carcinoma cell proliferation and this effect is achieved through reduced oxygen consumption accompanied by ROS formation [
309]. In addition, administration of a TPP
+ derivative of chlorambucil (Mito-Chlor) causes an 80-fold increase in cell killing in breast and pancreatic cancer cell lines and delays tumour progression in a mouse xenograft model of human pancreatic cancer [
310]. TPP
+ was also conjugated to doxorubicin (DOX) to generate TPP-DOX derivative. Enhanced cytotoxicity and apoptosis were observed for TPP-DOX compared to free doxorubicin in the MDA-MB-435 cancer cell line. These effects were more pronounced in doxorubicin-resistant than in doxorubicin-sensitive cells, suggesting that preferential distribution of doxorubicin to the mitochondria can revert drug resistance in tumour cells [
311].
In addition to TPP
+, a couple of other DLCs (either alone or attached to the spacer) have already been used to target various anti-cancer pharmacophores into mitochondria in order to destabilize them in cancer cells, with F16, rhodamine (B and 101), rhodacyanine MKT-077, dequalinium, heterocyclic aromatic cations, natural and synthetic mitochondria-targeting peptides and mitochondria-targeted nanoparticle (NP) vesicles being the most important ones. Both natural and synthetic compounds have functionally been conjugated to these DLCs. As a consequence, the resulting biological effects of the conjugates usually significantly exceed those of their precursors. As an example, F16-conjugated natural pentacyclic triterpenoids showed a considerable enhancement of antitumour action in comparison with the parent compounds and a markedly higher cytotoxic effect against tumour cell lines over healthy fibroblast cells [
312]. The same findings were reported for triterpenoids linked to TPP
+ [
313,
314,
315,
316,
317]. Pentacyclic triterpenoid-conjugates with rhodamine also displayed increased cytotoxic effects in cancer cells compared to non-malignant fibroblasts [
318,
319].
Although detailed information on how DLC conjugates mediate mitochondria-targeted cytotoxic effects that are highly cancer-specific is still missing, a couple of mechanisms have been suggested. Depending on the particular DLC compound and its conjugate, these include a surface-active effect on mitochondrial membranes causing organelle aggregation, a dose-dependent decrease in mitochondrial transmembrane potential, suppression of oxidative phosphorylation, an increase in H
2O
2 generation, induction of apoptosis in an ROS-mediated manner, suppression of the STAT3 activation pathway, autophagy, cell cycle arrest, inhibition of mtDNA replication, inhibition of the activity of complexes of the respiratory chain, influence on the balance between pro- and anti-apoptotic proteins and others (for more details, see [
320,
321]).
Despite the increasing number of anti-cancer drugs used for treatment of solid tumours, CDDP remains a widely used conventional therapy. However, the application of CDDP has numerous limitations, including drug resistance and off-target effects. To overcome these limitations, derivatives of this drug have been designed, synthetized and tested. Among them, monofunctional CDDP complexes represent a highly promising class of such derivatives, in which lonidamine (an inhibitor of mitochondrial hexokinase) is anchored to the CDDP centre for the selective de-energization of cancer cells. One such complex among the monofunctional Pt(II) complexes—monofunctional Pt(III) (MPL-III)—was reported to be more potent than CDDP in an MDA-MB-231 TNBC cell line, although it exhibited relatively low cytotoxicity towards breast epithelial cells. The MPL-III derivative was further shown to mainly accumulate in the mitochondria, where it induced detrimental changes to the mitochondrial ultrastructure, caused significant loss of the MMP, inhibited glycolysis and disrupted mitochondrial respiration. Consequently, MPL-III caused cell cycle arrest in the G0/G1 phase and mitochondria-mediated apoptosis involving caspase activation and cytochrome c release. At the molecular level, MPL-III was found to perturb DNA damage repair pathways, metabolic processes and transcription regulation [
322]. Another way of overcoming CDDP resistance through mitochondrial targeting can be achieved with dinuclear Ir-CDDP complexes, in which an iridium(III) moiety is introduced to a terpyridyl CDDP derivative. The resulting compound, Ir-Pt, has exhibited a significant increase in mitochondrial accumulation and strong anti-tumour activity towards CDDP-resistant lung adenocarcinoma cells. This compound has been shown to severely damage mtDNA, disrupting the mitochondrial function, leading to loss of the MMP and depletion of ATP and resulting in cell death by necrosis [
323,
324].
Application of NPs provides a platform for more efficient ways to target mitochondria. For development of effective NP-based therapies, there is a substantial need to choose the right combination of mitochondria-penetrating peptides (MPPs) and cell-penetrating peptides (CPPs). To attack mtDNA with CDDP more efficiently using NPs, a hydrophobic mitochondria-targeting CDDP prodrug, Platine-M, was constructed through cycloaddition of an azide-Pt(IV) precursor to TPP
+-bound azadibenzocyclooctyne. Platine-M loaded onto the surface of a biocompatible triblock polymeric NP accumulated effectively inside the mitochondrial matrix of chemoresistant ovarian carcinoma cells, which was accompanied by significantly better Platin-M activity than pure CDDP [
325]. Another approach to target mitochondria of cancer cells using specific peptides as the basis for CDDP delivery involves tethering of
cis-ammineplatinum(II) complex [Pt(succac)(NH
3)
2](NO
3) to the N terminal end of the MPPs supplemented by unnatural amino acids, particularly D-arginine and L-cyclohexylalanine [
326]. Interestingly, the linkage of Pt(IV) prodrug with oligonucleotide gold nanoparticle conjugates (DNA-AuNP) increased cytotoxicity compared to CDDP in human lung carcinoma A549, human prostate cancer PC3 and cervical cancer HeLa cell lines [
327]. Short interfering RNAs, particularly silencing RNAs (siRNAs), conjugated with CPP (or other carriers) enable systemic delivery of siRNA into the cell, where it may induce sequence-specific posttranscriptional modifications in targeted transcripts crucially affecting further progress of cancer cells [
328]. As an example, mesoporous silica nanoparticles (MSNs) loaded with siRNA targeting the protooncogene
BCL-2 along with a chemotherapeutic drug in the NP core showed a synergistic effect against chemoresistant TNBC [
329]. Another dual drug conjugate composed of α-tocopheryl succinate (α-TOS) and CDDP, or, alternatively, doxorubicin or paclitaxel, blended with lipids and polyethylene glycol using a lipid-film hydration method was examined in the HeLa cell line. α-TOS elevated the effect of each individual drug, which was demonstrated by both nDNA damage and cytochrome c release from mitochondria [
330]. In addition, targeting mitochondrial complex II of ETC with α-TOS conjugated to TPP
+ increased the permeability of the resulting TOS-TPP
+-obatoclax (BCL-2 inhibitor) NP in the outer mitochondrial membrane, resulting in apoptosis of TNBC cells [
331]. Human breast cancer cell lines MCF-7 and T47D were successfully targeted by a covalent conjugate composed of nucleolin, the peptide targeting breast cancer cell receptor, and thiol groups containing α-lipoic acid when both were loaded onto an AuNP a stable receptor-specific peptide-AuNP was formed [
332]. Eventually, mitochondria could be targeted with a dual drug conjugate composed of CDDP and the 3-bromopyruvate inhibitor that affects glycolytic enzyme hexokinase 2 within OXPHOS [
325].
All the above examples prove that mitochondria indeed provide a suitable environment that may be therapeutically targeted to further improve cancer therapy. A number of studies have demonstrated how targeting different aspects of mitochondrial processes can successfully contribute to cancer treatment. However, the biggest challenge in the development of effective anti-cancer drugs is to create pharmacologic regulators respecting the multifaceted nature of the individual processes participating in tumorigenesis in each specific cancer type or subtype. Nevertheless, among the different therapeutic approaches, mitochondria-targeted DNA damaging agents still possess high potency and the ability to better evade therapy resistance mechanisms and off-target effects, combating the cancer cells through mechanisms that are distinct from those achieved by free drugs.