Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marcela Káňová | -- | 3539 | 2022-08-09 17:17:44 | | | |
| 2 | Catherine Yang | + 1 word(s) | 3540 | 2022-08-10 03:41:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kanova, M.; Kohout, P. Intensive Care Unit-Acquired Weakness and Sarcopenia. Encyclopedia. Available online: https://encyclopedia.pub/entry/25995 (accessed on 22 July 2026).
Kanova M, Kohout P. Intensive Care Unit-Acquired Weakness and Sarcopenia. Encyclopedia. Available at: https://encyclopedia.pub/entry/25995. Accessed July 22, 2026.
Kanova, Marcela, Pavel Kohout. "Intensive Care Unit-Acquired Weakness and Sarcopenia" Encyclopedia, https://encyclopedia.pub/entry/25995 (accessed July 22, 2026).
Kanova, M., & Kohout, P. (2022, August 09). Intensive Care Unit-Acquired Weakness and Sarcopenia. In Encyclopedia. https://encyclopedia.pub/entry/25995
Kanova, Marcela and Pavel Kohout. "Intensive Care Unit-Acquired Weakness and Sarcopenia." Encyclopedia. Web. 09 August, 2022.
Copy Citation
Intensive care unit-acquired weakness (ICUAW) brings about skeletal muscle wasting due to critical illness and has important clinical implications, significantly impacting rehabilitation, and increasing both morbidity and mortality. ICUAW is sometimes referred to as critical illness polyneuromyopathy—being called critical illness polyneuropathy (CIP) when nerve involvement predominates, or critical illness myopathy (CIM) where muscle involvement is crucial. It manifests as muscle weakness that develops rapidly, prior to any detectable muscle wasting.
intensive care unit-acquired weakness
sarcopenia
proteostasis
ubiquitin–proteasome system
1. Early Muscle Weakness in ICUAW
Muscle weakness manifests very early, especially in sepsis patients with the development of ICUAW. This weakness is seen long before the researchers are able to detect any loss of muscle mass (cross-sectional area), or loss of muscle protein through inflammation-mediated proteolysis. Mechanisms contributing to muscle weakness include impaired intracellular Ca2+ homeostasis resulting in reduced fiber contractility, mitochondrial dysfunction with bioenergetic failure, channel dysfunction with membrane inexcitability, and hyperglycaemic toxicity [1][2].
1.1. Caspases, Calpains, and the Disruption of Myofilament Structure
Functionally, there are two types of muscle fibers. Slow-twitch muscle fibers, known as type I or red fibers (high blood supply) contain more mitochondria and myoglobin. They are aerobic, fatigue-resistant, and are focused on postural control. Fast-twitch muscle fibers—type II or white, anaerobic fibers provide a more powerful force, but over a shorter duration and fatigue quickly. These type II fibers are more sensitive to the multifactorial injury caused by critical illness.
The integrity of contractile myofilaments and the regular striated pattern of actin and myosin are essential for sufficient muscle strength. Caspase and calpain initially disrupt the structure of actin and myosin myofibrils, which reduce contractility with the early loss in force generation [3][4]. These proteases disrupt myofilament structure by cleaving actin and myosin from sarcomeres, and supply proteins to the UPS, which is activated by pro-inflammatory cytokines. Tumor necrosis factor (TNF) and NFκB accelerate ubiquitination: the process of marking proteins for degradation.
Caspases are active not only in the initiation of protein breakdown by UPS, but also in apoptosis (caspase3). Calpains are calcium-dependent cysteine proteases that are involved in the function of the UPS (m and μ calpains), with calpain3 (also known as p94) being specific to muscle. It plays an important role in activated protein degradation during sepsis. It is possible that disruption of myofilament structure contributes to the early loss in force generation [3].
Sarcopenia largely affects fast-twitch muscle fibers. Reduction in muscle mass and strength leads to impaired mobility, along with an increased risk of falls, compromised independence, and lowered quality of life [5].
1.2. Decrease in Calcium Release from the Sarcoplasmic Reticulum
The function of calpains is calcium-dependent, as is the activation of ubiquitination. Thus, calcium regulates protein breakdown. Above all, it is essential for muscle contraction, has a direct effect on myosin ATPase, and influences glycolysis and oxidative metabolism. Calcium homeostasis is ensured in the sarcoplasmic reticulum. Outflow of Ca2+ through ryanodine receptors is ATP-dependent. ATP deficiency reduces Ca2+ release from the sarcoplasmic reticulum, affecting membrane excitability of skeletal muscle. Force generation is, thus, rapidly reduced during sepsis [2][4][6][7].
1.3. Sodium Channels and Electrical Inexcitability
Excitation is necessary to generate an action potential, which leads to changes in the permeability of the membrane to sodium, potassium, and possibly calcium ions. Following excitation, the resting energy potential (−80 to −90 mV) increases to reach the threshold potential at which sodium channels open. Na+ thus enters the intracellular space, and the inner side of the membrane becomes increasingly positively charged compared to the outer side of the membrane, resulting in depolarization (+20 to +30 mV). Potassium channels then open and K+ leaks out of the cell to maintain electroneutrality, and membrane voltage decreases, causing repolarization. In septic patients, alterations to membrane sodium pumps can cause disturbances in electrical excitability. Inflammatory cytokines have a neurotoxic effect, causing chronic membrane depolarization, functionally manifesting as “denervation”. The resting membrane potential of muscle fibers is reduced and is unable to reach the action potential, as voltage-gated ion channels follow an all-or-nothing rule. Thus, the rapid development of muscle weakness is initially more of a functional issue, affecting both nerves and muscles [3][8][9].
2. Mitochondrial Dysfunction
2.1. Muscle Mass, and Polyneuromyopathy in the Critically Ill
Sufficient energy is a prerequisite for muscle contraction; therefore, mitochondria, the organelles that are the “power plant” of the organism, are central to muscle function.
Mitochondria are subcellular organelles that furnish the cell with adenosine triphosphate (ATP), which they generate by oxidative phosphorylation, and are absolutely essential for the energetic processes of every cell. In addition to this extremely important function, they are involved in calcium homeostasis and intracellular reactive oxygen species (ROS) generation, mediate intracellular communication, and regulate apoptosis. Mitochondrial damage is associated with a lack of released energy (ATP), ROS overproduction, and the release of cytochrome c. Ultrastructural damage to the mitochondria and mitochondrial dysfunction also contribute to organ failure [10][11].
Mitochondria are sensitive to oxidative damage. On the one hand, mitochondria are the main source of free radicals, and on the other hand, are a key target for oxidative damage. Oxidative stress is typical during sepsis, a dysregulated response to severe infection, but it is also quite common during aging. Senescence is accompanied with a progressively increasing accumulation of ROS in line with the gradual decline in the capacity of the antioxidant defense system. When the defense system is no longer able to cope with the enhanced rate of oxidant production, cellular and subcellular environments become more susceptible to damage. Mitochondrial dysfunction has been demonstrated to activate cell apoptosis and can ultimately result in organ damage [12][13].
Mitochondrial dysfunction is, thus, a key player in the development of ICUAW in the context of critical illness, most progressively in sepsis. Age-related mitochondrial dysfunction plays an equally important role in the development of sarcopenia with the progressive decline in mitochondrial bioenergetics. The typical manifestation is a reduction in maximal oxygen uptake (VO2 max) and a consequent decrease in exercise tolerance [11][14].
Further, alteration of mitochondrial permeability transition pores (mPTP) is one of the mechanisms of sarcopenia, and results in ROS overproduction, and triggers muscle atrophy by activating the FoXO transcription factor family and ubiquitin ligases, thus activating proteolysis. Open mPTP can lead to the release of pro-apoptotic factors and released cytochrome c can increase proteasomal activity [15].
2.2. Mitochondria in Skeletal Muscle
Skeletal muscle enables movement and maintains posture, but it also has a role in thermoregulation, ensuring nutritional balance, glucose uptake, and is a hormone source [16][17]. Myokines are released from muscle during exercise; they are proteins with endocrine and paracrine functions that control inflammatory processes, angiogenesis, and myofibril hypertrophy, and also modulate fuel oxidation [16].
Skeletal muscle is a highly energetic tissue that typically contains mitochondria in three locations: subsarcolemmal, perinuclear, and intermyofibrillar. While subsarcolemmal mitochondria provide resistance to ROS, intermyofibrillar mitochondria are the source of ATP during oxidative phosphorylation and Ca2+ modulation. With regard to metabolism, the energy source in red myofibers (type I) is slow oxidative (SO) aerobic oxidation. Fast (or white) muscle fibers use glycolysis as the energy source, with fast oxidative and glycolytic reactions (FOG) in type IIa, and only fast glycolytic (FG) reactions in type IIb fibers [11].
2.3. Mitochondrial Quality Control Mechanisms
Mitochondria have to adapt rapidly to changing energy requirements, energy supply from aerobic or anaerobic pathways, and oxidative stress. This plasticity of mitochondria in response to energy demand is regulated by multiple molecular signals. This regulation happens in stages ranging from mitochondrial biogenesis, through mitochondrial dynamics (fusion and fission), to mitochondrial autophagy (mitophagy). A reduced capacity for mitochondrial quality control leads to mitochondrial dysfunction—a key marker of multiple organ failure. Skeletal muscles are among those that are affected early and significantly [10].
2.3.1. Mitochondrial Biogenesis
Mitochondrial biogenesis is the process by which new mitochondria are produced. The main regulator of mitochondrial biogenesis is proliferator-activated receptor γ coactivator 1α (PGC-1α). This interacts with other transcription factors such as FoXO, hepatocyte nuclear factor (HNF 4α), or nuclear respiratory factor (NRF1, NRF2), and activates transcription from replication of MtDNA (Figure 1).
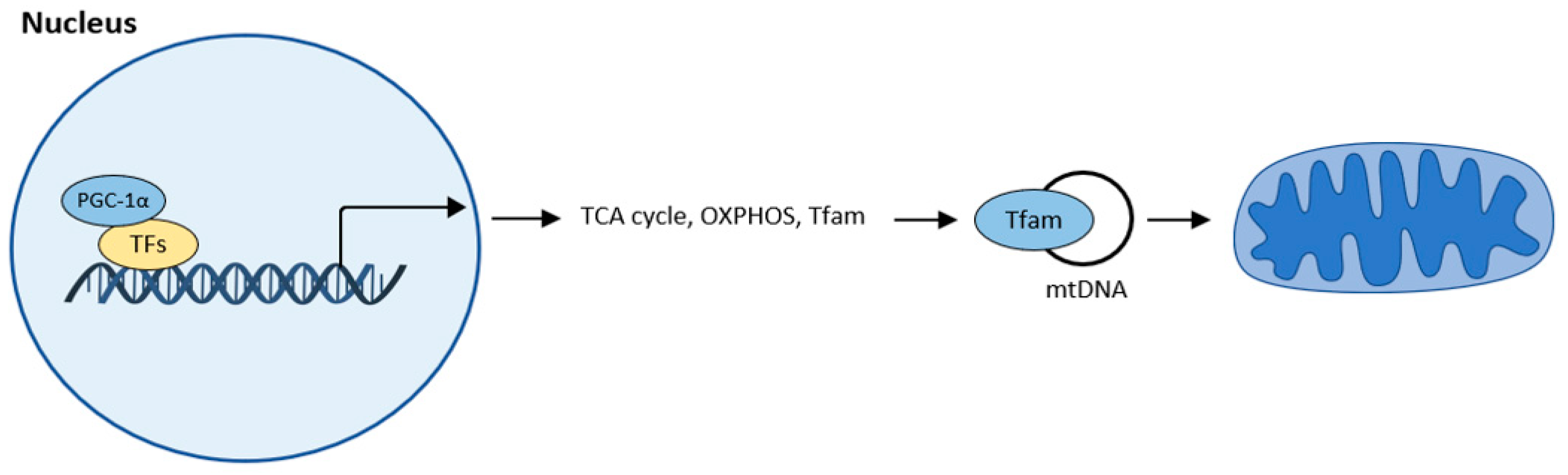
Figure 1. Mitochondrial biogenesis according to [10]. The central regulator of mitochondrial biogenesis: peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Transcription factors (TFs): forkhead box class-O (FoxO1), hepatocyte nuclear factor 4a (HNF4a), nuclear respiratory factor (NRF1, NRF2).
2.3.2. Mitochondrial Dynamics
Mitochondria adapt their number, morphology, and size to the energy requirements and can either fuse or divide by fission under the influence of GTPase. GTPases controlling mitochondrial fission include the fission protein dynamin-related protein1 (Drp1) and its receptor mitochondrial fission protein (Fis1), the mitochondrial fission factor (Mff), and mitochondrial dynamics proteins (MiD49, MiD51). Fusion is controlled by mitofusin (Mfn) and optic atrophy 1 (OPA 1) (Figure 2) [15][18][19][20][21].
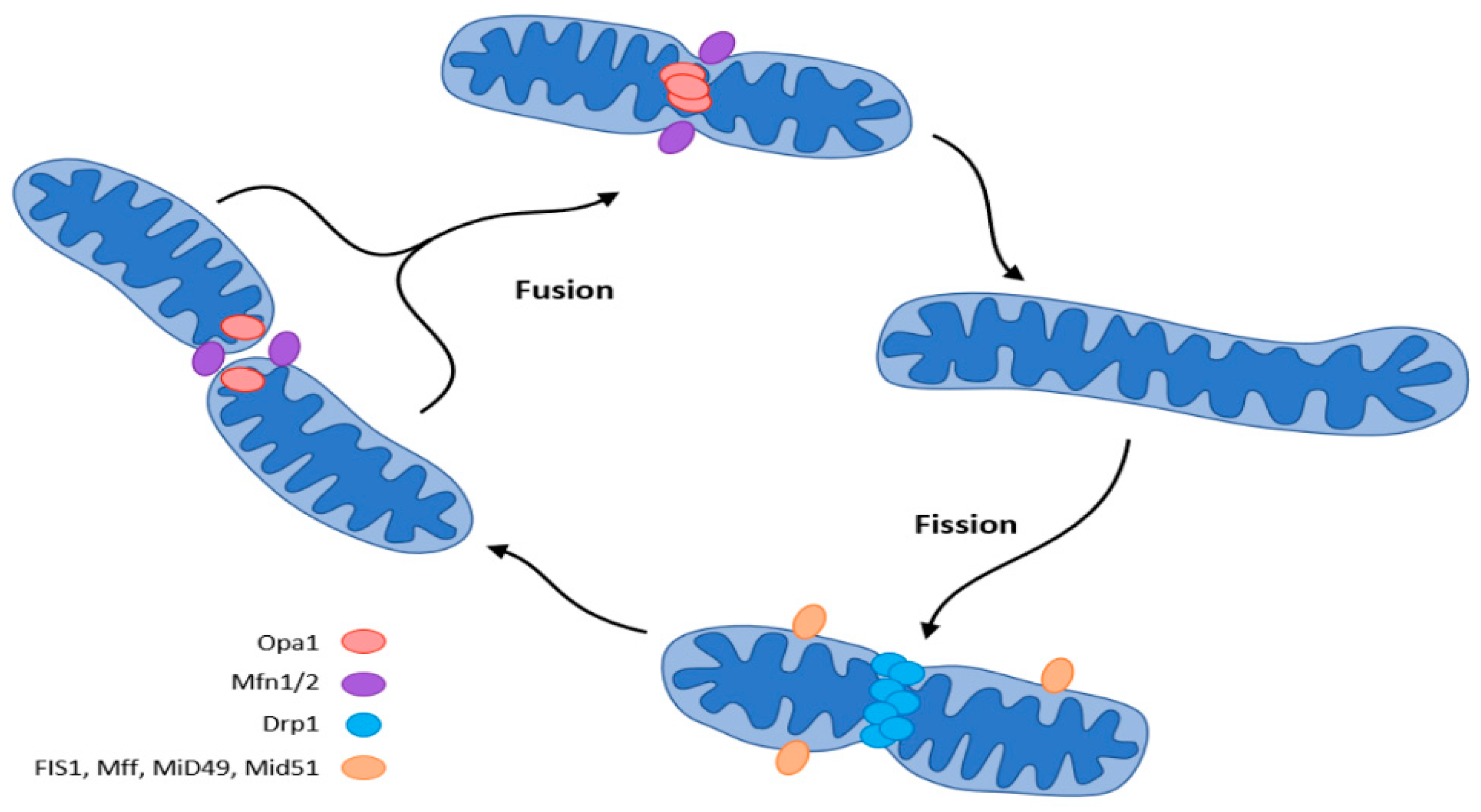
Figure 2. Mitochondrial dynamics (fission and fusion events) according to [15]. The central regulator of mitochondrial biogenesis: peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α). Transcription factors (TFs): forkhead box class-O (FoxO1), hepatocyte nuclear factor 4a (HNF4a), nuclear respiratory factor (NRF1, NRF2).
2.3.3. Mitochondrial Autophagy
The final step in quality control is mitochondrial autophagy or mitophagy. This process eliminates damaged organelles by forming a double-membrane autophagosome, which then fuses with the lysosome where the contents are degraded. This requires receptors that recognize damaged mitochondria, where the main regulator of mitophagy is Pink1/Park2. Pink1 (PTEN-induced putative protein kinase) and Park2 act as an E3 ubiquitin ligase. In healthy mitochondria, Pink1 moves from its position in the outer membrane to the inner mitochondrial membrane and when its N-terminal mitochondrial targeting sequence (MTS) is exposed to the matrix, it is recognized by matrix processing peptidase (MPP) that cleaves the p64 form of Pink1 to its p53 form, which is subsequently released into the cytosol and cleaved by the proteasome. Thus, Pink1 does not activate autophagy in healthy mitochondria.
This is not the case in damaged mitochondria, where sepsis results in the loss of mitochondrial membrane potential. Pink1 does not move to the inner membrane but is sequestered in the outer membrane of the mitochondria, and subsequently recognized and labeled by the bound ubiquitin ligase Park2. Mfn (mitochondrial fusion protein) is also labeled and subsequently cleaved by the proteasome, preventing mitochondrial fusion. Park2 builds ubiquitin chains that can be sufficient to recruit autophagic receptors such as protein 62 (p62), nuclear dot protein (NDP52), optineurin, and others to initiate autophagosome formation, thus triggering mitophagy. This allows damaged mitochondria to be removed. Inhibition of this “scavenging” action leads to the progression of MODS, thereby increasing mortality. The accumulation of damaged mitochondria has shown to trigger motor neuron and muscle fiber degeneration, and upon progression of muscle dysfunction, can lead to ICUAW or sarcopenia [10][22][23][24] (Figure 3).
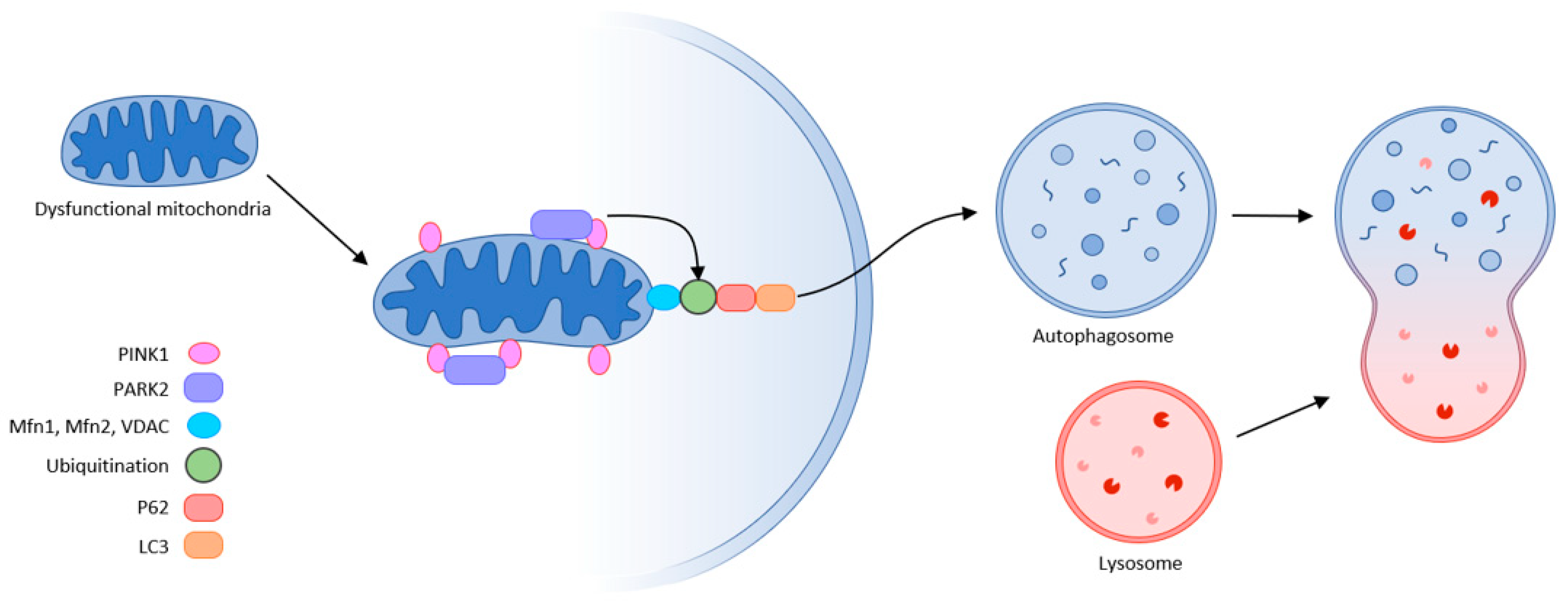
Figure 3. Mitophagy (schematic representation of the autophagy machinery) partly according to [10].
3. Potential Targets for Intervention in ICUAW
There are no interventions that can consistently treat ICUAW or sarcopenia; therefore, most interventions focus on reducing or eliminating risk factors. Nonetheless, several new potential targets of signaling pathways are currently being evaluated in clinical trials.
3.1. Mitochondrial Monitoring and Therapy
Monitoring mitochondrial dysfunction is already possible using PCR on mitochondrial DNA (MtDNA). A new highly sensitive, quantitative droplet method can monitor MtDNA copies in stimulated peripheral blood mononuclear cells. Reduction in their levels is a sign of oxidative stress both in critical illness and in aging, where they are a sign of frailty [11][25].
Oxidative stress (ROS) and nitric oxide (NO) inhibit electron transport chains and leads to mitochondrial swelling. The current approach is to modulate action at the subcellular level. Activation of mitochondrial biogenesis is used for mitochondrial therapy; in sepsis, this effort to recover mitochondrial function can prevent organ failure. L-carnitine, succinate, ATP-MgCl2, cytochrome c, and ubiquinol (CoQ) all have the same effect. Inhaled CO can rescue liver failure in sepsis (by inducing heme oxygenase-1-mediated NF-E2-related factor 2) and CoQ can prevent LPS-induced mitochondrial dysfunction by improving mitochondrial biogenesis [11][26][27]. Bezafibrate, a drug used for dyslipidemia, is an agonist of peroxisome proliferator-activated receptor (PPAR) and can increase PGC-1α expression [28]. Similarly, the oral antidiabetic drug metformin activates mitochondrial biogenesis. Glutathione and melatonin can be targeted as mitochondrial antioxidants [11].
LPS disrupts mitochondrial physiology in skeletal muscle via its pleiotropic effects on sphingolipid metabolism. Treatment with myocrin, a de novo sphingolipid biosynthesis inhibitor, ameliorates skeletal muscle dysfunction by decreasing sepsis-induced Drp1 expression and subsequently reverting mitochondrial morphology, inhibiting excessive mitochondrial fission and restoring the balance between fusion and fission [22]. However, therapeutic intervention on mitochondrial dynamics aimed at suppressing muscle atrophy and the development of sarcopenia—e.g., by suppressing Drp1 overexpression to attenuate the aging-related accumulation of mitochondrial dysfunction and sarcopenia—can have conflicting results. The network of signaling cascades is very dense with many feedback loops. Mitochondrial fission must be maintained to ensure mitochondrial and muscle health [29].
Impaired mitophagy also contributes to amplifying organ failure in sepsis. Targeting this scavenging process is another option for mitochondrial therapy. Carbamazepine, lithium, and sodium valproate are used as autophagic flux enhancers. Rapamycin and activated protein C are pharmacological agents used to induce autophagy. However, rapamycin affects many other metabolic pathways (e.g., proteosynthesis through the mTOR system) [30]. Current nutritional recommendations for critically ill patients are based on this principle. Hypocaloric nutrition with a gradual increase in energy and protein is recommended at the beginning so that autophagy does not become “excessive” [25]. Adenosine monophosphate-activated protein kinase (AMPK) and the silent mating-type information regulation 2 homolog sirtuin (SIRT1) are two of the best-known metabolic sensors that can directly affect PGC-1α sensitivity and mitochondrial biogenesis [30].
Moderate long-term exercise stimulates metabolic adaptations in aged skeletal muscle through the activation of PGC-1α, AMPK, and the SIRT1 pathway. Endurance exercise not only stimulates mitochondrial biogenesis but also causes repeated oxygen free radical loading. This results in an increase in antioxidant capacity. In addition to ROS production during exercise, myofibrils are damaged and, thus, proteolysis is stimulated after exercise with an increase in the amino acid pool and subsequent stimulation of proteosynthesis, thus increasing muscle mass and strength. Regular exercise tends to maintain low levels of oxidative damage and improve proteostasis, preventing sarcopenia [11][31][32]. Blood flow-restricted exercise (BFR) using a pneumatic tourniquet system in working musculature results in inadequate oxygen supply. BRF mediates muscle hypertrophy through protein signaling and satellite cell proliferation to a greater extent than resistance exercise alone [2][33]. The critically ill and often geriatric sarcopenic patients are unable to exert sufficient training loads to increase muscle mass. Neuromuscular electrical stimulation shows positive results for preventing skeletal muscle weakness and wasting in critically ill patients [34].
3.2. The Critical Nature of the Proteolytic System
Muscle is metabolically very active and is constantly challenged by mechanical, oxidative, and heat stresses. A properly functioning proteolytic system is essential to maintain proper function and tissue recovery. Improper proteolysis leads to myopathies; given this picture of the negative impact of proteolysis, its original objective of adaptation to stress is lost. Activated skeletal muscle proteolysis and its effect on the development of polyneuromyopathy in critically ill patients can be used in efforts to suppress proteolysis, but proper timing of proteasome inhibition is therefore crucial [35].
Skeletal muscle cell (myoblast) differentiation depends on the early activation of appropriate myogenic factors. UPS is involved in cell differentiation, initially removing the paired-box transcription factors Pax3 and Pax7, thereby initiating the transformation of satellite cells. Its role is critical for the early activation of the key myogenic factor MyoD by removing its inhibitor Id, and other myogenic factors requiring proteolytic cleavage such as E2A proteins, filaminB, Myf5, and myogenin. This initiates the transformation of the satellite cells first to myoblasts and then to myotubes. All the ROS generated during the entire process of myogenesis also need to be scavenged [36][37].
During muscle hypertrophy following resistance exercise, both proteosynthesis and degradation of damaged proteins are increased, and UPS (through the proteasome axis (MuRF1—muscle-specific RING finger protein1, MAFbx—atrogin1)) facilitates myofilament restructuring and growth [38].
Caspases, besides initiating apoptosis, have an important role in skeletal myoblast differentiation. They activate satellite cells following the cleavage of the promyogenic kinases MST1, HIPK2, NEK5, and CAD and the cleavage of Pax7 (caspase3). Caspase3 enhances myoblast fusion, a critical step in muscle maturation [36][39]. As it is one of the major protein degradation pathways, exacerbated autophagy can induce ICUAW. On the other hand, “basal” autophagy is necessary to maintain muscle mass and prevent muscle atrophy. It has an important role in preventing sarcopenia, where stem cell senescence limits muscle regeneration in aging. Without autophagy, efficient clearance of damaged organelles and proteins would not work during muscle stress adaptation following exercise or starvation. Exercise can activate beclin1 (by phosphorylation and release from the BCL2–beclin1 complex), allowing autophagosome formation. The extent of activation of autophagic signaling is influenced by the duration and intensity of exercise. Autophagy is activated during exercise in ultra-endurance runners; the highly increased skeletal muscle expression of key autophagy genes such as Atg4b, Atg12, LC3, Bnip3, etc., is necessary for the removal of dysfunctional proteins and to meet the elevated energetic demands. FOXO3 phosphorylation decreases at the same time as proteasome activation. So, balanced autophagy is necessary to maintain normal protein turnover [2][36] (Figure 4).
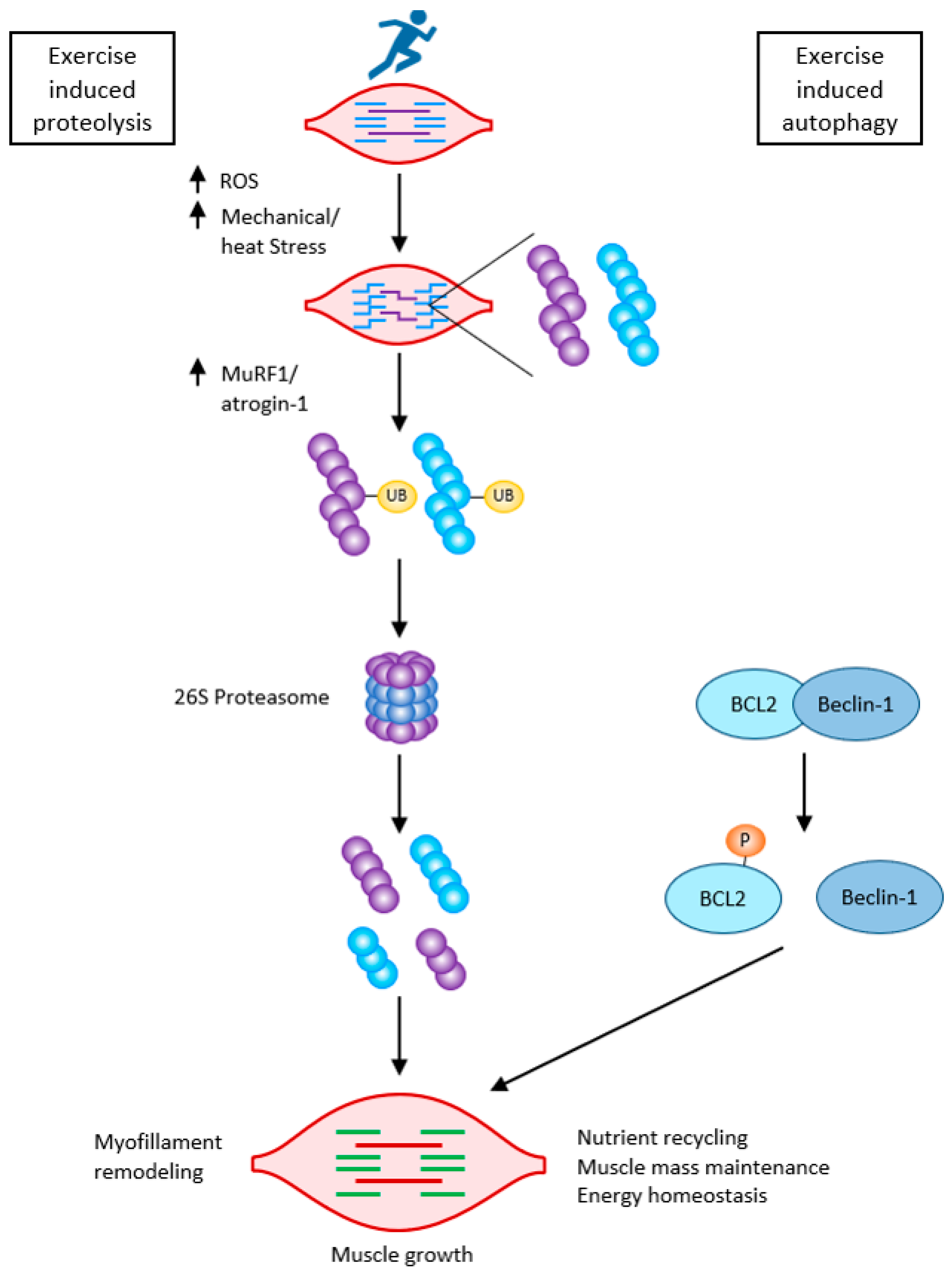
Figure 4. Exercise-induced muscle growth through induced proteolysis and induced autophagy according to [36]. Exercise-induced protein damage via increased ROS/mechanical and heat stress, increased MuRF1, and atrogin-1 (MAFbx), and both muscle-specific ubiquitin ligases lead to the activation of the 26 S proteasome to rid the cells of non-functional myofibrillar proteins. Exercise also activates autophagy: beclin-1 is phosphorylated and released from the BCL2–beclin-1 complex. Exercise-induced autophagy is necessary for the clearance of damaged organelles and proteins. This is critical for skeletal muscle remodeling and growth.
3.3. The Role of Starvation and Stress Metabolism
Nutritional supplementation plays an important role in the prevention of ICUAW and the development of sarcopenia, especially protein sufficiency, in an attempt to influence the link between proteosynthetic mTOR and proteolytic UPS. The main stimulators of mTOR include branched-chain amino acids, especially leucine and its metabolite hydroxymethylbutyrate (HMB) [40]. Leucine uses both insulin-dependent (IGF-IR/Akt/mTORC1) and insulin-independent (RagD/mTORC1) pathways to stimulate mTOR. This latter pathway of activation via Ragulator is common to both leucine and its metabolite HMB. Other amino acids use other activation pathways, e.g., arginine and glutamine use the lysosomal-sensing protein SLC38A9/mTORC1 [41].
An important role is also played by sufficient vitamin D, considered to be a steroid hormone. Vitamin D receptors (VDR) are found in various tissues, and are also presented on skeletal muscle, predominantly in fast-twitch muscles. Vitamin D is associated with oxidative stress, muscle energy metabolism, mitochondrial functions, and acts in Ca2+ homeostasis, which is necessary for muscle contraction. By reducing Ca2+ reuptake into the sarcoplasmic reticulum, it prolongs the relaxation phase of muscle contraction. Vitamin D deficiency decreases protein synthesis (through a signaling cascade: decreased IGF-1/Akt/mTOR), and on the other hand, decreased IGF-1/Akt activates FoXO and triggers muscle atrophy by elevation of MuRF1 and atrogin-1. UPS is also stimulated via attenuation of steroid receptor coactivator complex (Src) and decreased PGC-1α. The availability of vitamin D decreases with age, so it has a role in sarcopenia. Supplementation of vitamin D, to a minimum level of 75–100 nmol/L (30–40 ng/mL), reduces the symptoms of myopathy. Of course, these problems cannot be fully solved with vitamin D alone; what is important is bioavailability and the right level [42][43]. Nonetheless, robust evidence for this is still lacking.
3.4. The Role of Glucocorticoids
Systemic inflammation and elevation of cytokines (TNF, IL1, IL6) drive muscle atrophy. They activate the mitogen-activated protein kinase (MAPK) and upregulate atrogin-1 (MAFbx) and MuRF1.
Glucocorticoids are commonly used in patients to suppress cytokine storms in critically ill patients, including COVID-19 patients [44]. However, they have a markedly negative impact on muscle dysfunction, activating UPS with the concomitant increase in atrogin-1 and MuRF1. Therapeutically, glucocorticoid receptor blockade can be used to manage glucocorticoid-induced skeletal muscle atrophy. This may attenuate UPS activated by acidosis, insulin resistance, or sepsis. There are numerous inhibitors to block MAFbx and/or MuRF1, including eIF3-f, MyoD, and myogenin. However, this therapy has had indifferent success, possibly because of the different causes of atrophy, multiple signaling pathways, and no one specific inhibitor for all stages of atrophy [45][46]. Nevertheless, IL1 blockade with anakinra has shown admirable results in survival of sepsis patients [47][48][49].
3.5. Specifics of Therapy for Sarcopenia
Therapies for sarcopenia, or efforts to slow muscle loss due to aging, rely on sufficient intake of quality protein (leucine, phenylalanine, and arginine—with new recommendations to increase protein intake up to 1.5 g/kg/day), limitation of saturated fats, and sufficient omega3 fatty acids and vitamin D, along with exercise. Active lifestyle and exercise can improve repair myogenesis and increase the expression of neuronal form of NOS in the muscle, thus leading to the activation of satellite cells (SC). SC are located between the basal lamina and sarcolemma of muscle fibers; they are the principal contributors to muscle repair and growth and decrease significantly during aging. Moreover, myostatin, an extracellular messenger of the transforming growth factor superfamily, inhibits factors that regulate myogenesis. Exercise has been observed to decrease this muscle growth-inhibiting messenger. During exercise, increased levels of vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF) modulate oxidative stress, improve brain-derived neurotrophic factor (BDNF), and thus improve stimulation of skeletal muscle contraction [50][51][52][53][54].
Several clinical studies have explored possible pharmacological interventions in signaling pathways to manage sarcopenia. Among them are myostatin inhibitors (Bimagrumab and the more recent trevogrumab and domagrozumab). Ghrelin, important for food intake with anabolic properties, is being tested for appetite reduction and malnutrition. ACE inhibitors, such as perindopril or the selective AT inhibitor losartan, are also known to be beneficial by increasing IGF-1 levels [55].
References
- Kress, J.P.; Hall, J.B. ICU-acquired weakness and recovery from critical illness. N. Eng. J. Med. 2014, 370, 17.
- Lad, H.; Saumur, T.M.; Herridge, M.S.; Cdos Santos, C.; Mathur, S.; Batt, J.; Gilbert, P.M. Intensive Care Unit-Acquired Weakness: Not just another muscle atrophying condition. Int. J. Mol. Sci. 2020, 21, 7840.
- Bloch, S.; Polkey, M.I.; Griffiths, M.; Kemp, P. Molecular mechanisms of intensive care unit-acquired weakness. Eur. Respir. J. 2012, 39, 1000–1011.
- Zink, W.; Kaess, M.; Hofer, S.; Plachky, J.; Zausig, Y.A.; Sinner, B.; Weigand, M.A.; Fink, R.H.; Graf, B.M. Alteration in intracellular Ca2+ homeostasis of skeletal muscle fibers during sepsis. Crit. Care Med. 2008, 36, 1559–1563.
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Writing Group for the European Working Group on Sarcopenia in Older People 2 (EWGSOP2), and the Extended Group for EWGSOP2. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 601.
- Clark, M.G.; Rattigan, S.; Barrett, E.J. Nutritive blood flow as and essential element supporting muscle anabolism. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 185–189.
- Friedrich, O.; Hund, E.; Weber, C.; Hacke, W.; Fink, R.H.A. Critical illness myopathy serum fractions affect membrane excitability and intracellular calcium release in mammalian skeletal muscle. J. Neurol. 2004, 251, 53–65.
- Khan, J.; Harrison, T.; Rich, M. Mechanisms of neuromuscular dysfunction in critical illness. Crit. Care Clin. 2008, 24, 165–177.
- Z’Graggen, W.J.; Lin, C.S.Y.; Howard, R.S.; Beale, R.J.; Bostock, H. Nerve excitability changes in critical illness polyneuromyopathy. Brain 2006, 129, 2461–2470.
- Wu, J.; Yao, Y.M.; Lu, Z.Q. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J. Mol. Med. 2019, 97, 451–462.
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of age-related mitochondrial dysfunction in sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236.
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64.
- Boengler, K.; Kosiol, M.; Mayr, M.; Schulz, R.; Rohrbach, S. Mitochondria in ageing. Role in heart, skeletal muscle and adipose tissue. J. Cachexia Sarcopenia Muscle 2017, 8, 349–369.
- Short, K.R.; Vitone, J.L.; Bigelow, M.L.; Proctor, D.N.; Nair, K.S. Age and aerobic exercise training effects on whole body and muscle protein metabolism. Am. J. Phyisiol. Endocrinol. Metab. 2004, 286, E92–E101.
- Leduc-Gaudet, J.P.; Hussain, S.N.A.; Barreiro, E.; Gouspillou, G. Mitochondrial dynamics and mitophagy in skeletal muscle health and aging. Int. J. Mol. Sci. 2021, 22, 8179.
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195.
- Guidicde, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55.
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256.
- Kalia, R.; Wang, R.Y.R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405.
- Otera, H.; Wang, C.X.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158.
- Song, Z.Y.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 Mediate Sequential Steps in Mitochondrial Membrane Fusion. Mol. Biol. Cell 2009, 20, 3525–3532.
- Hansen, M.E.; Simmons, K.J.; Tippetts, T.S.; Thatcher, M.O.; Saito, R.R.; Hubbard, S.T.; Trumbull, A.M.; Parker, B.A.; Taylor, O.J.; Bikman, B.T. Lipopolysaccharide disrupts mitochondrial physiology in skeletal muscle via disparate effects on sphingolipid metabolisms. Shock 2015, 44, 585–592.
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314.
- Calvani, R.; Joseph, A.M.; Adhihetty, P.J.; Miccheli, A.; Bossola, M.; Leeuwenburgh, C.; Bernabei, R.; Landi, F.; Marzetti, E. Update on mitochondria and muscle aging: All wrong roads lead to sarcopenia. Biol. Chem. 2018, 399, 421–436.
- O’Hara, R.; Tedone, E.; Ludlow, A.; Huang, E.; Arosio, B.; Mari, D.; Shay, J.W. Quantitative mitochondrial copy number determination using droplet digital PCR with single-cell resolution. Genome Res. 2019, 29, 1878–1888.
- MacGarvey, N.C.; Suliman, H.B.; Bartz, R.R.; Fu, P.; Withers, C.M.; Welty-Wolf, K.E.; Piantadosi, C.A. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor 2 induction rescue mice from lethal Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 2012, 185, 851–861.
- Bullon, P.; Roman-Malo, L.; Marin-Aguilar, F.; Alvarez-Suarez, J.M.; Giampieri, F.; Battino, M.; Cordero, M.D. Lipophilic antioxidants prevent lipopolysaccharide-induced mitochondrial dysfunction through mitochondrial biogenesis improvement. Pharmacol. Res. 2015, 91, 1–8.
- Hondares, E.; Pineda-Torra, I.; Iglesias, R.; Staels, B.; Villarroya, F.; Giralt, M. PPARdelta, but not PPARalpha activates PGC-1alpha gene transcription in muscle. Biochem. Biophys. Res. Commun. 2007, 354, 1021–1027.
- Dulac, N.; Leduc-Gaudet, J.P.; Cefis, M.; Ayoub, M.B.; Reynaud, O.; Shams, A.; Moamer, A.; Nery Fereira, M.F.; Hussain, S.N.; Gouispillou, G. Regulation of muscle and mitochondrial health by the mitochondrial fission protein Drp1 in aged mice. J. Physiol. 2021, 599, 4045–4063.
- Yen, Y.T.; Yang, H.R.; Lo, H.C.; Hsieh, Y.C.; Tsai, S.C.; Hong, C.W.; Hsieh, C.H. Enhancing autophagy with activated protein C and rapamycin protects against sepsis-induced acute lung injury. Surgery 2013, 153, 689–698.
- Van Zanten, A.R.; De Waele, E.; Wischmeyer, P.E. Nutritional therapy and critical illness: Practical guidance for the ICU, post-ICU, and long-term convalescence phases. Crit. Care 2019, 23, 368.
- Musci, R.V.; Hamilton, K.L.; Linden, M.A. Exercise-Induced Mitohormesis for the maintenance of skeletal muscle and the health span extension. Sport 2019, 7, 170.
- Patterson, S.D.; Hughes, L.; Warmington, S.; Burr, J.; Scott, B.R.; Owens, J.; Abe, T.; Nielsen, J.L.; Libardi, C.A.; Laurentino, G.; et al. Blood flow restriction exercise: Consideration of methodology, application, and safety. Front. Physiol. 2019, 10, 533.
- Maffiuleti, N.A.; Roig, M.; Karatzanos, E.; Nanas, S. Neuromuscular electrical stimulation for preventing skeletal-muscle weakness and wasting in critically ill patients: A systematic review. BMC Med. 2013, 11, 137.
- Hermans, G.; Van den Berge, G. Clinical review: Intensive care unit acquired weakness. Critical Care 2015, 19, 274.
- Bell, R.A.V.; Al-Khalaf, M.; Megeney, L.A. The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet. Muscle 2016, 6, 16.
- Hass, K.F.; Woodruff, E.; Brodie, K. Proteasome function is required to maintain muscle cellular architecture. Biol. Cell 2007, 99, 615–626.
- Murton, A.J.; Constantin, D.; Greenhaff, P.L. The involvement of the ubiquitin proteasome system in humans skeletal muscle remodeling and atrophy. Biochim. Biophys Acta 2008, 1782, 730–743.
- Larsen, B.D.; Rampalli, S.; Burns, L.E.; Brunette, S.; Dilworth, F.J.; Megeney, L.A. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc. Natl. Acad. Sci. USA 2010, 107, 4230–4235.
- Suryawan, A.; Rudar, M.; Fioroto, M.L.; Davis, T.A. Differential regulation of mTORC1 activation by leucine and β-hydroxy-β-methylbutyrate in skeletal muscle of neonatal pigs. J. Appl. Physiol. 2020, 128, 286–295.
- Tan, V.P.; Miyamoto, S. Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals. J. Mol. Cell Cardiol. 2016, 95, 31–41.
- Dzik, K.P.; Kaczor, J.J. Mechanisms of vitamin D on skeletal muscle function: Oxidative stress, energy metabolism and anabolic state. Eur. J. Appl. Physiol. 2019, 119, 825–839.
- Sinha, A.; Hollingsworth, K.G.; Ball, S.; Cheetham, T. Improving the vitamin D status of vitamin D deficient adults is assocciated with improved mitochondrial oxidative function in skeletal muscle. J. Clin. Endocrinol. Met. 2013, 98, E509–E513.
- Qin, E.S.; Hough, C.L.; Andrews, J.; Bunnell, A.E. Intensive care unit-acquired weakness and the COVID-19 pandemic: A clinical review. PM R 2022, 14, 227–238.
- Folleta, V.C.; White, L.J.; Larsen, A.E.; Léger, B.; Russel, A.P. The role and regulation of MAFbx/atrogin-1 and MuRF1 in skeletal muscle atrophy. Eur. J. Physiol. 2011, 461, 325–335.
- Combaret, L.; Taillandier, D.; Darvedet, D.; Bechet, D.; Rallière, C.; Claustre, A.; Grizard, J.; Attaix, D. Glucocorticoids regulate mRNA levels for subunits of the 19 S regulatory complex of the 26 S proteasome in fast-twitch skeletal muscle. Biochem. J. 2004, 378, 239–246.
- Hughes, L.; Paton, B.; Rosentblatt, B.; Gissane, C.; Patterson, S.D. Blood flow restriction training in clinical musculoskeletal rehabilitation: A systemic review and metaanalyses. Br. J. Sports Med. 2017, 51, 1003–1011.
- Shakoory, B.; Carcillo, J.A.; Chatham, W.W.; Amdur, R.L.; Zhao, H.; Dinarello, C.A.; Cron, R.Q.; Opal, S.M. Interleukin-1 receptor blockade is associated with reduced mortality in sepsis patients with features of macrophage activation syndrome. Crit. Care Med. 2016, 44, 275–281.
- Kooistra, E.J.; Waalders, N.J.B.; Grondman, I.; Janssen, N.A.F.; de Nooijer, A.H.; Netea, M.G.; van de Veerdonk, F.L.; Ewalds, E.; van der Hoeven, J.G.; Kox, M.; et al. Anakinra treatment in critically ill COVID-19 patients: A prospective cohort study. Crit. Care 2020, 24, 688.
- Fernández, J.P.; Montero, A.F.; Matínez, A.C.; Pastor, D.; Rodrígez, A.M.; Roche, E. Sarcopenia: Molecular pathways and potential targets for intervention. Int. J. Mol. Sci. 2020, 21, 8844.
- Kim, J.S.; Cross, J.M.; Bamman, M.M. Impact of resistence loading on myostatin expression and cell cycle regulation in young and older men and women. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1100–E1119.
- Paproski, J.J.; Finelo, G.C.; Murillo, A.; Mandel, E. The importance of protein intake and strength exercises for older adult. JAAPA 2019, 32, 32–36.
- Abiri, B.; Vafa, M. The role of nutrition in attenuating age-related skeletal muscle atrophy. Adv. Exp. Med. Biol. 2020, 1260, 297–318.
- Martínez-Arnau, F.M.; Fonfría-Vivas, R.; Buigues, C.; Castillo, Y.; Molina, P.; Hoogland, A.J.; vanDoesburg, F.; Pruimboom, L.; Fernández-Garrido, J.; Cauli, O. Effects of leucine administration in sarcopenia. A randomized and placebo-controlled clinical trial. Nutrients 2020, 12, 932.
- Yoon, J.H.; Kwon, K.S. Receptor-mediated muscle homeostasis as a target for sarcopenia therapeutics. Endocrinol. Metab. 2021, 36, 478–490.
More
Information
Subjects:
Critical Care Medicine
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.5K
Entry Collection:
Peptides for Health Benefits
Revisions:
2 times
(View History)
Update Date:
10 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No