Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anikó Borbás | -- | 3655 | 2022-08-04 16:10:20 | | | |
| 2 | Dean Liu | Meta information modification | 3655 | 2022-08-05 03:06:52 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bege, M.; Borbás, A. The Medicinal Chemistry of Artificial Nucleic Acids. Encyclopedia. Available online: https://encyclopedia.pub/entry/25861 (accessed on 28 July 2026).
Bege M, Borbás A. The Medicinal Chemistry of Artificial Nucleic Acids. Encyclopedia. Available at: https://encyclopedia.pub/entry/25861. Accessed July 28, 2026.
Bege, Miklós, Anikó Borbás. "The Medicinal Chemistry of Artificial Nucleic Acids" Encyclopedia, https://encyclopedia.pub/entry/25861 (accessed July 28, 2026).
Bege, M., & Borbás, A. (2022, August 04). The Medicinal Chemistry of Artificial Nucleic Acids. In Encyclopedia. https://encyclopedia.pub/entry/25861
Bege, Miklós and Anikó Borbás. "The Medicinal Chemistry of Artificial Nucleic Acids." Encyclopedia. Web. 04 August, 2022.
Copy Citation
Nucleic acids play a central role in human biology, making them suitable and attractive tools for therapeutic applications. While conventional drugs generally target proteins and induce transient therapeutic effects, nucleic acid medicines can achieve long-lasting or curative effects by targeting the genetic bases of diseases.
oligonucleotides
phosphorothioate
phosphorodiamidate morpholino oligomers (PMOs)
peptide nucleic acids
xenonucleic acids with neutral backbone
xenonucleic acids with cationic backbone
base modifications
sugar modifications
2'-substituted nucleic acids
loc
1. Artificial Nucleic Acids
The therapeutic use of natural nucleic acids is hampered by their sensitivity to nucleases and their poor pharmacological properties due to their polyanionic nature. These dificulties can be overcome by proper chemical modifications to either the nucleobase, the ribofuranose unit or the internucleotide linkage [1]. Nuclease stability can be enhanced by modifying the phosphodiester backbone and sugar units. Backbone modifications also modulate pharmacokinetic properties including cellular uptake. Sugar modifications may enhance hybridization efficiency, which can be quantitatified by Tm value, i.e., the temperature at which the two strands of duplex are separated. Hybridization selectivity (Tm decrease/mismatch) can be improved by altering the nucleobases or the backbone.
Before discussing the chemical structure and biological properties of XNAs, researchers briefly describe the process of oligonucleotide synthesis. During biosynthesis, enzymes build up nucleic acids from the 5′-end monomer to the 3′-end. The chemical synthesis of oligonucleotides and artificial nucleic acids—which is generally performed by automated solid phase oligonucleotide synthesis (SPOS)—proceeds in the opposite direction. In this case, the first nucleoside is attached to the solid phase, which is usually a controlled pore glass (CPG), through its 3′-OH group, while the 5′-hydroxyl is protected in form of a 4,4′-dimethoxytrityl (DMTr) ether. Then, the synthesis cycle consisting of four reactions is started. First, DMTr group is cleaved with trichloroacetic acid (TCA) to release the 5′-OH group. In the next step, 3′-O-(N,N-diisopropyl)phosphoramidite derivative of 5′-O-DMTr-nucleoside is added together with the activating agent which is an acidic azole catalyst, e.g., 1H-tetrazole or 4,5-dicyanoimidazole. In this coupling step a phosphite triester internucleotide linkage is formed. Subsequently, due to the incompleteness of the coupling, a capping is necessary, which means the protection of the remaining 5′-hydroxyl groups by acetylation with Ac2O in order to exclude the unreacted monomers from further synthetic cycles. In the last step of the cycle, the phosphite derivative obtained is oxidized into a phosphate with iodine. The above steps must be repeated n-2 times to obtain an oligomer of n nucleoside in length. Finally, the protecting groups are removed and the complete oligomer is cleaved from the solid phase. Benzoyl group is used to protect the amino groups of the nucleobases in cytidine and adenosine, and i-butyryl is used to protect the amino group of guanosine. In RNA synthesis, the 2′-OH group is protected with tert-butyldimethylsilyl ether (TBDMS) (Scheme 1) [2][3][4][5]. In addition to the above typical protocol, there are other methods for SPOS. The synthesis of modified nucleic acids may involve other steps or modifications.

Scheme 1. Solid-phase synthesis of oligonucleotides by phosphoramidite chemistry. The iterative steps of synthetic cycle are highlighted in red. DMTr: 4,4′-dimethoxytrityl, TBDMS: t-butyldimethylsilyl, TCA: trichloroacetic acid, Ac2O: acetic anhydride, CPG: controlled pore glass.
2. Base Modified Nucleic Acids
Instead of canonical bases such as thymine/uracil, adenine, cytosine and guanine, other bases can be incorporated into nucleic acids that can can be involved in hibridization (Figure 1). G-clamps (9-(2-aminoethoxy)-phenoxazine derivatives) are tricyclic nucleobase analogues named after the G-shaped device of the same name. They are functionally cytosine analogues, because they can bind to guanine, however G-clamp can form four hydrogen bonds with guanine, instead of the 3 H-bond in the G-C pair. This greatly increases the binding strength of the G-clamp-containing oligonucleotides and may increase the efficacy of hybridization [6][7][8]. Pseudoisocytosine may be part of a Hoogsteen base pair without protonation, therefore pseudoisocytosine-containing oligonucleotides have good triplex-forming potential, for example with DNA during anti-gene gene silencing, therefore, pseudoisocytosine is favourably used in peptide-nucleic acids (vide infra) [9][10].
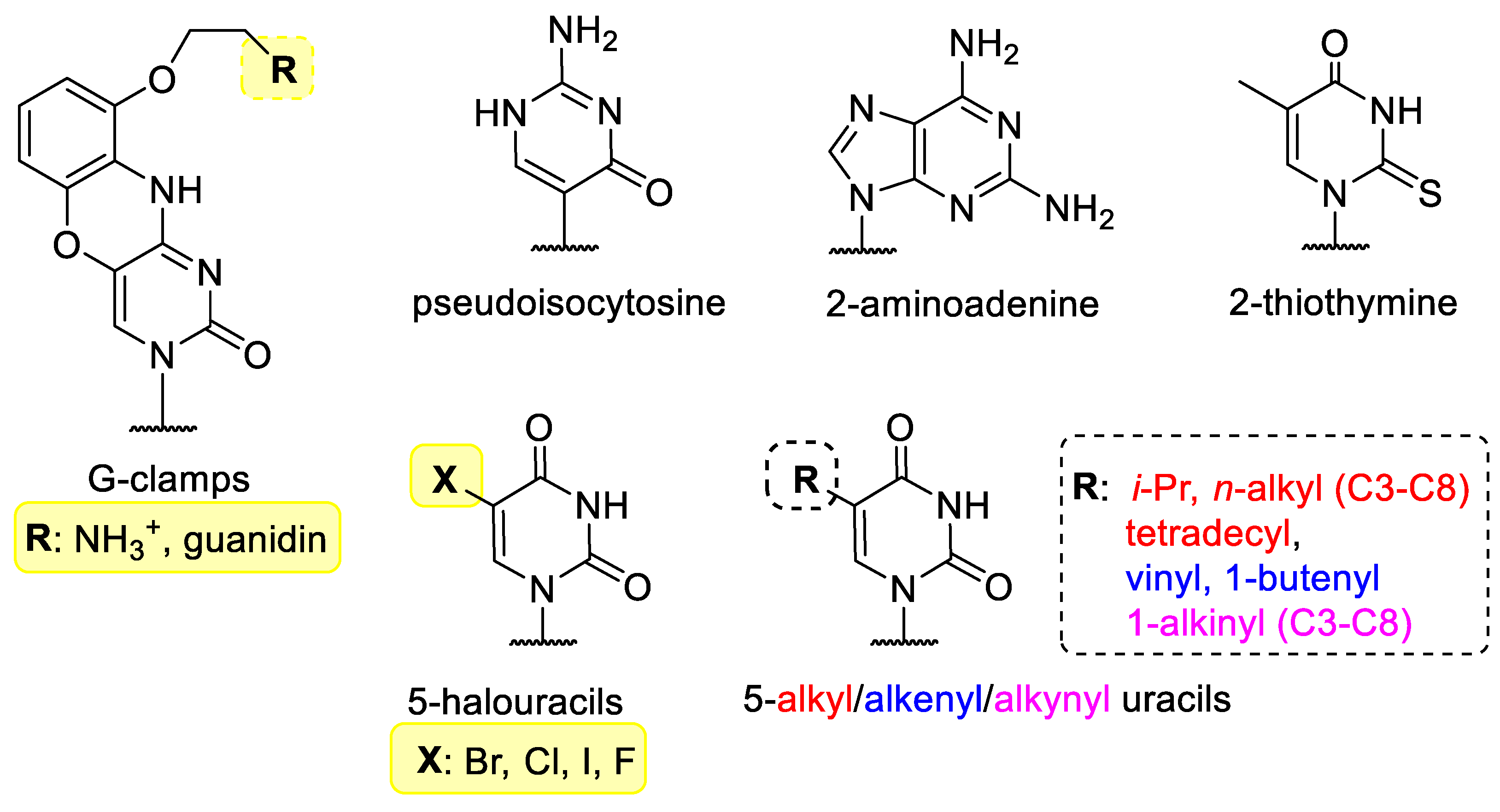
Figure 1. Modified bases in XNAs.
2-Aminoadenine and 2-thiothymine bind weakly to each other, but can bind strongly to the natural nucleobases [11][12]. The 5 position of pyrimidine nucleobases is not involved in hybridization and is therefore often chosen to incorporate halogens [13] or small alkyl, alkenyl or alkinyl groups [14].
3. Sugar-Modified Nucleic Acids
Selected examples for sugar-modified nucleosides are shown in Figure 2. The β-D-ribofuranose unit of natural NAs can be replaced by α or L stereoisomers or other sugars. L-nucleotides are enantiomers of D-forms, therefore their oligomers are called “spiegelmers” (spiegel is mirror in German). Spiegelmers are resistant to nucleases and are not immunogenic [15]. They are not used in gene silencing, but have great potential as aptamers [16].
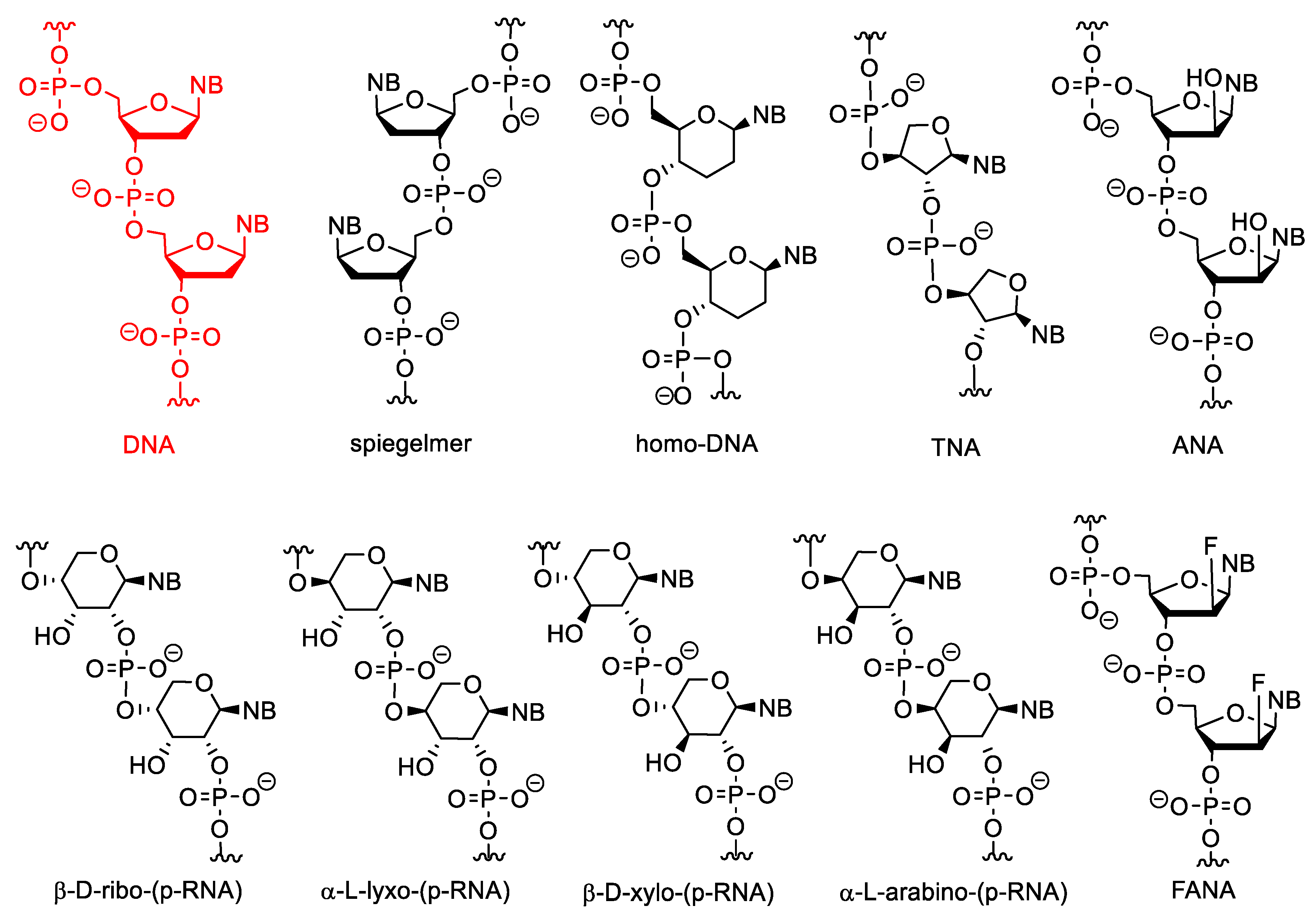
Figure 2. The structure of DNA and XNAs with other sugars than β-D-ribofuranose.
Nucleic acids may be composed of α-nucleotides, and these derivatives may have strong antisense activity [17][18]. The α and L nucleotides can be combined with other modifications, such as “locking” (see below) [19].
In pyranosyl RNAs (p-RNA), the ribofuranose ring is replaced by pento- or hexopyranoses. These derivatives have been synthesized primarily not for therapeutic purposes but as tools to study the relationships between the structure and biological functions of nucleic acids and thus to better understand the structural criteria of natural nucleic acids [20][21][22][23]. HomoDNA is an NA analogue that contains 2′,3′-dideoxyglucopyranose instead of ribose (altro-, and allopyranose also have been investigated). Base pairs between homoDNAs do not closely follow the Watson–Crick rules, A-A and G-G base pairs can also be formed according to antiparallel reverse Hoogsteen pairing [24].
Pentopyranosyl oligonucleotides (p-RNAs) contain a pentopyranose ring (β-D-ribose, β-D-xylose, α-L-lyxose or α-L-arabinose) as the sugar component, as the name implies. Base-pairing of these derivatives is not only stronger but even more selective than in DNA or RNA. p-RNAs are able to hybridize with each other [25][26][27][28]. TNAs (threofuranosyl nucleic acids) contain α-L-threofuranose instead of β-D-ribofuranose. Unlike β-pyranose nucleic acids, TNAs can hybridize to DNA and RNA. The hybridization strength of TNA duplexes is similar to that of the corresponding DNA or RNA suplexes [29]. HomoDNAs are connected through a 6′→4′ linkage, whereas 4′→2′ and 4′→3′ derivatives were formed in p-RNA. In TNA the phosphodiester bonds are in the 3′→2′ position. In arabinonucleic acids (ANA), the configuration of the C-2′ position is changed. These derivatives can form duplexes with RNA and DNA and activate RNAse H. Interestingly, the conformation of the sugar ring (“pucker”) in these molecules is closer to DNA than to RNA [30]. Their 2′-deoxy-2′-fluoro analogues are FANAs, which form more stable duplexes with RNA and especially with DNA [31].
Cyclohexanyl nucleic acids (CNA) contain cyclohexane ring (Figure 3). Both D and L isomers were synthesized and investigated. The Tm of D-CNA/D-CNA and L-CNA/L-CNA duplexes is higher than that of the L-CNA/D-CNA duplexes. Only D-CNA can hybridize to RNA and DNA [32]. Their 5′-6′ unsaturated analogues are the cyclohexenyl nucleic acids (CeNA). Incorporating cyclohexenyl monomers into DNA oligonucleotides, slightly decreases the Tm value of the duplex formed with complementary DNA but increases the Tm of the duplex, formed with complementary RNA [33]. The CeNA-RNA duplexes can activate RNAse H [34]. Hexitol nucleic acids (HNAs) contain 1,5-anhydrohexitol. All four isomers (β-D, α-D, β-L, α-L) were synthesized, however β-D-HNA was the first and most studied [35]. They can hybridize to natural nucleic acids, especially RNA [36] and can be used for gene silencing in antisense strategy [37] or RNA interference [38]. α-D and α-L-HNA can also form duplexes with RNA, while β-L-HNA is unable to do this [35].
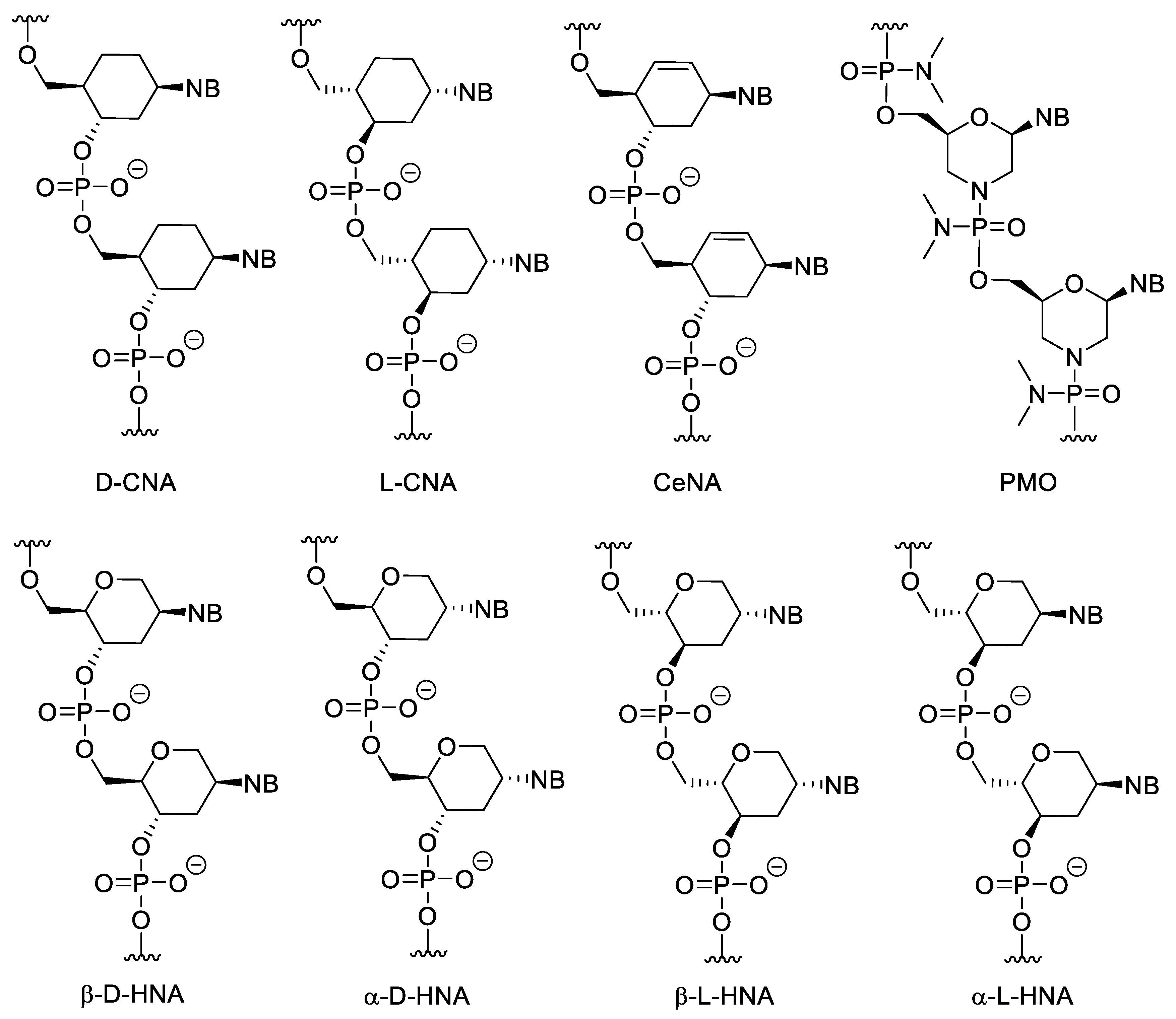
Figure 3. Structure of CNAs, CeNA, HNAs and PMO. PMOs contain both sugar and backbone modifications.
Phosphorodiamidate morpholino oligomers (PMOs) contain modifications to both the sugar unit and the backbone. The furanose units are replaced with morpholine rings attached to each other through phosphorodiamidate linkages. The morpholine motif is obtained by oxidizing the corresponding ribonucleoside derivative into a 2′,3′-secodialdehyde with IO4− and then forming the nitrogen-containing ring by a reductive amination-cyclization reaction. PMOs have several advantages, such as good water solubility, inexpensive synthesis, high stability in biological systems due to their resistance to many hydrolytic enzymes, good hybridization properties and high specificity [39]. Due to its neutral backbone PMO is less prone to bind to proteins than native NAs, which decreases non-specific, undesirable interactions. Importantly, PMO/RNA duplexes cannot activate RNase H. In gene silencing, this may be a disadvantage because the level of the targeted mRNA is not decreased by enzymatic degradation. However, this property can be exploited. Since the formed duplexes are not cleaved, non-targeted but non-specifically binding nucleic acids do not degrade, which means a higher selectivity. In addition, due to the lack of the degradation, PMOs can be used to modulate the splicing of pre-mRNA, to gain alternatively spliced mRNAs, which can be therapeutically used. Their low cellular uptake can be improved by conjugating them to cell penetrating peptides (CPPs) or guanidin containing moieties [40][41]. PMOs can be used as gene silencing tools against viruses [40]. PMO mediated gene silencing has been experimented in, among other things, zebrafish [42], xenopus [43] and sea urchin [44]. There are a few approved PMO-based drugs on the market today (see more below).
Some sugar-modified nucleoside derivatives contain bi- or tricyclic groups instead of the monocyclic ribofuranose (Figure 4). In general, these compounds show higher conformational rigidity than the mono- or acyclic NA analogues. In bicyclo-DNA, a cyclopentane ring is condensed to a tetrahydrofuran, which is connected to the nucleobase in 1′-position, while the OH groups involved in the internucleotide phosphate-ester bond are in positions 3′ and 5′. The poly-T bicyclo-DNA binds to poly-A DNA weaker than a complementary non-modified DNA oligomer. However, poly-A bicyclo-DNA hybridize to the complementary poly-T DNA stronger, than the natural poly-A DNA. The Tm of poly-A/poly-T bicyclo-DNA is higher than that of the DNA/DNA duplex with the same sequence. Bicyclo-DNA prefers the Hoogsteen and reverse Hoogsteen base pairing over the Watson–Crick [45][46].
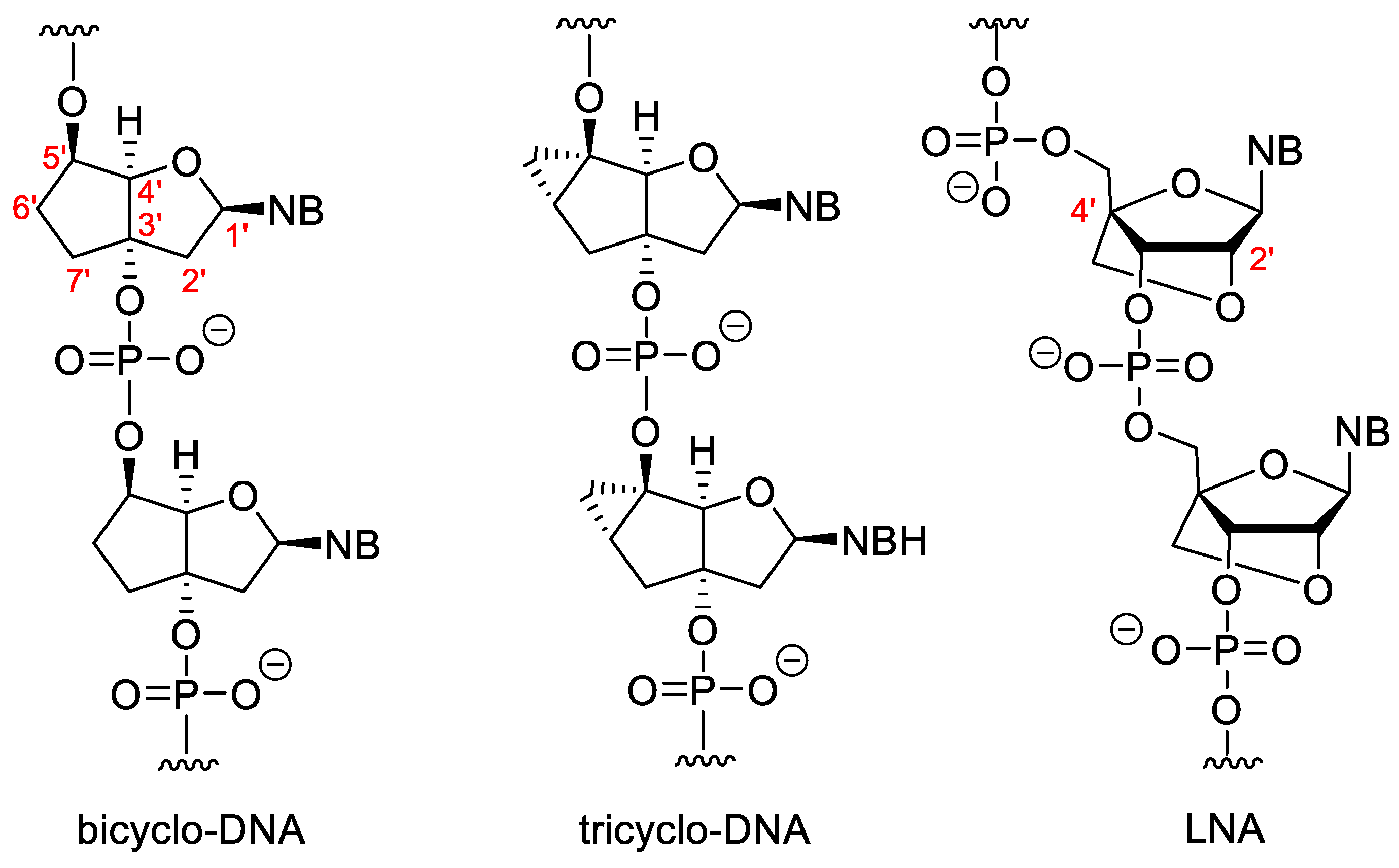
Figure 4. Bi- and tricyclic XNAs.
Tricyclo-DNAs (tc-DNA) are similar to the bicyclo analogues, but in this case a third cyclopropane ring is condensed to the cyclopentane. Tricyclo DNA can form stronger duplexes with complementary DNA and RNA than the non-modified DNA/DNA or DNA/RNA duplexes, and complementary tricyclo-DNAs form very stable duplexes with each other [47]. They are very effective antisense agents. They have high serum stability and are unable to activate RNAse H [48][49]. Except for the poly-A and poly-T sequences, tricyclo-DNA prefers Watson-Crick pairing [50].
In “locked” nucleic acids (LNAs) the 2′-O is attached to the 4′-C via a methylene group. These dervatives are also known as 2′,4′-bridged nucleic acids (BNA).The methylene bridge fixes the furanose ring in the 3′-endo conformation that is highly advantageous for base pairing. LNA forms very stable duplexes with RNA and DNA according to the Watson–Crick rule with excellent selectivity. The incorporation of LNA monomers into DNA oligomer increases the strength of hybridization [51]. There are modified LNAs with different configurations or that contain nitrogen or sulphur in the 2′-position instead of oxygen, but the “parent” β-D-LNA possesses the most advantageous properties [52].
Besides mono-, and multicyclic analogues, there are acyclic XNAs, which have a more flexible backbone (Figure 5). The general advantage of these derivatives is their relatively simple and cost-effective synthesis. In the unlocked nucleic acid (UNA) the covalent bond between the 2′ and 3′ carbons is removed. They can be obtained from ribonucleosides by oxidation to 2′,3′-secodialdehydes with metaperiodate followed by reduction of the formyl groups obtained into OH-s. The incorporation of UNA monomers into DNA or RNA destabilizes the duplexes. A more interesting phenomenon is where hybridization selectivity can be influenced by incorporation of UNA monomers at the appropriate points of the oligomer. If the mismatch is located distally to the position of the UNA unit, mismatch discrimination is increased. While in case of a proximal mismatch, the discrimination is decreased especially if the mismatch can be found directly opposite to the UNA unit. This effect can be used to adjust the selectivity of the hybridization of an oligomer [53]. Even the duplex destabilizing property can be exploited, since this way the undesirable binding of the XNA oligomer to nontargeted NAs can be reduced [54].
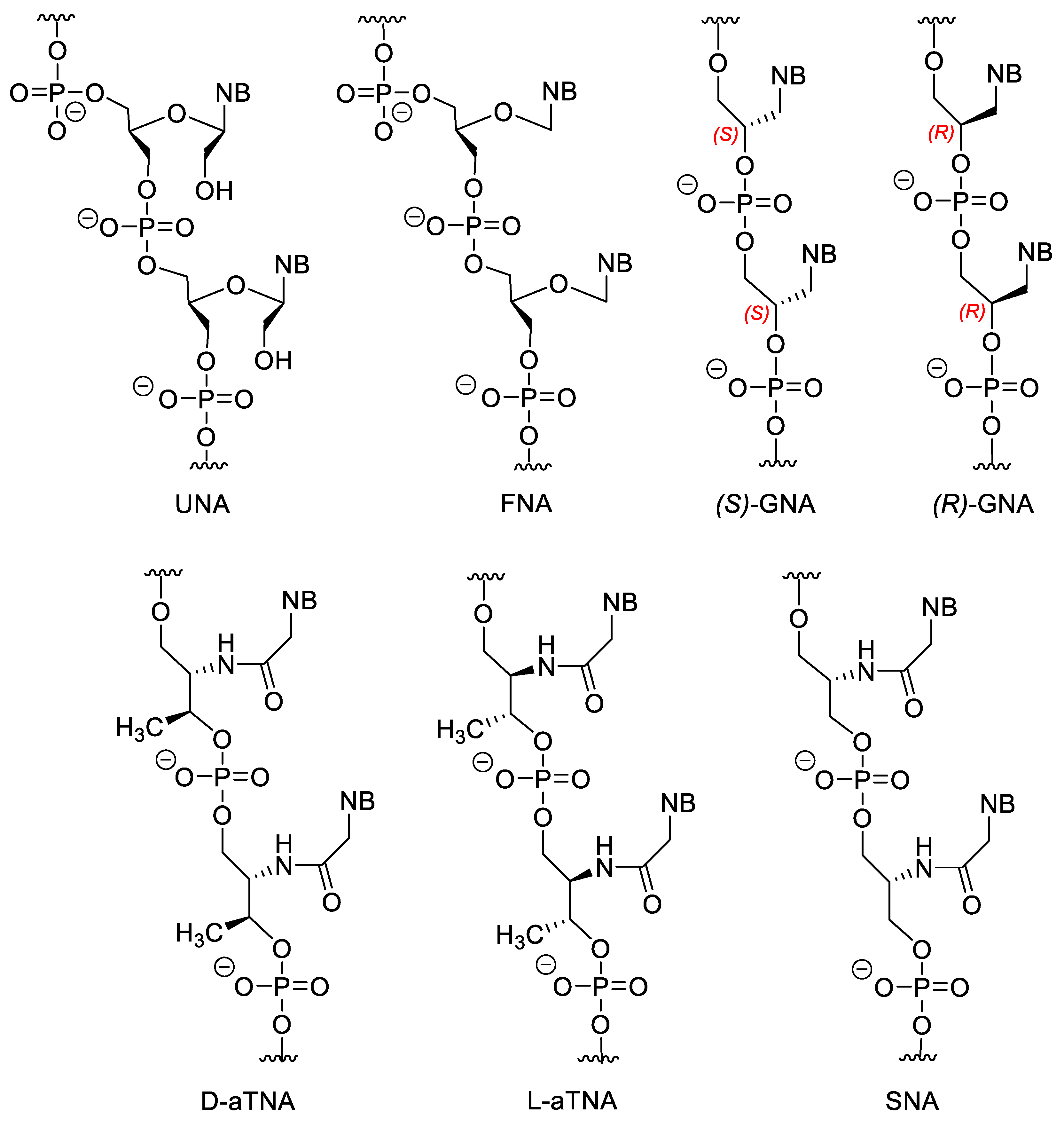
Figure 5. Acyclic NA analogues.
Flexible nucleic acid (FNA) does not contain the 2′-CH2OH group. FNA may weakly hybridize to DNA but, interestingely, FNA monomer triphosphates can be the substrates for DNA polymerase [55][56]. Theories suggest that the first information storing molecules of life could be similar molecules to FNA [57].
Both isomers of glycol nucleic acid (GNA) can form very stable homoduplexes, and (S)-GNA can hybridize to RNA, however, the latter depends highly on the sequence, high G/C ratio destabilize the GNA/RNA hybride [58][59].
There are also amino-acid-based XNAs. Acyclic threoninol nucleic acid contains D- or L-threoninol instead of ribose (D-aTNA and L-aTNA). D-aTNA can form very stable homoduplexes, but cannot hybridize to DNA or RNA. While L-aTNA can bind to DNA and RNA and form homoduplexes with similar stability to the D-isomer [60][61][62]. Serinol nucleic acids (SNA) can form duplexes with DNA and RNA [62][63]. Interestingly, the chirality of the SNA oligomer, depends only on the sequence. The enantiomer of an SNA oligomer is the reverse sequenced oligomer, therefore SNAs with a symmetric sequence are meso compounds.
Minor modifications can also be made on nucleosides (Figure 6). For example, the 2′-OH of ribose can be substituted with methyl, allyl, propyl, aminopropyl and other ether groups or can be replaced by a fluorine atom (2′-F RNA) [64]. Of these modifications, the 2′-O-methoxyethyl (MOE) substitution is the most important because the MOE nucleic acids have the most advantageous properties and this modification is common in second generation antisense oligonucleotide (ASO) drugs. The MOE modified nucleic acids have high binding strength and nuclease resistance [65][66][67]. However, importantly, fully 2′-modified ASOs cannot activate RNase H.
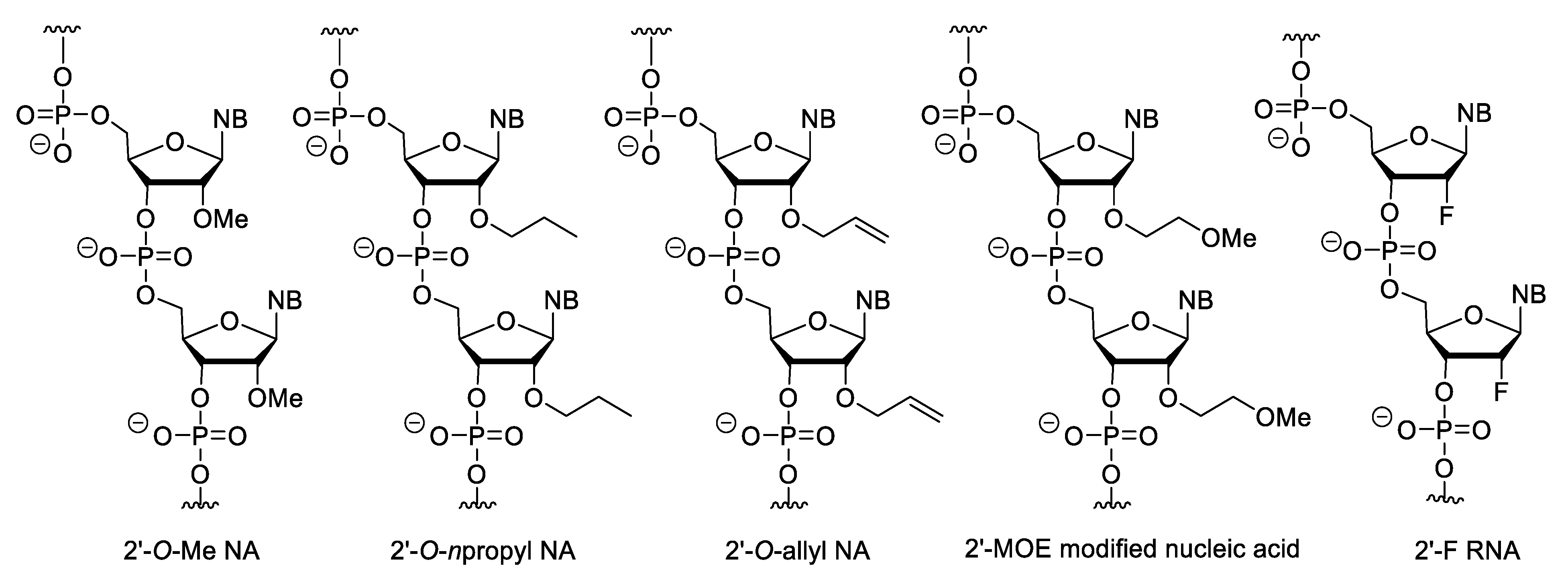
Figure 6. 2′-Substituted nucleic acid analogues.
4. Nucleotides with Modified Backbones
In phosphorothioate (PS) oligonucleotides, one of the non-bridging oxygen of the phosphate-ester group is changed to sulphur (Figure 7). This modification retains the negative charge of the backbone. They have highly increased nuclease resistance relative to the natural nucleic acids, and RNA/PS oligonucleotide duplexes can activate RNAse H [68]. Besides the above, PSs have the advantage, that they can be easily synthesized with conventional oligonucleotide synthesis methods with a minor modification of the protocol (exchanging the oxidation step for sulfur incorporation). However, during this method, a diastereoisomeric mixture is obtained (due to the chiral nature of the PS bond, there are two isomeric forms, the Rp or endo and the Sp or exo stereoisomers) [69]. Their disadvantages include that they form slightly less stable duplexes than unmodified NAs. In addition, due to their anionic backbone, they can non-specifically bind to proteins, which can cause unwanted biological effects [70]. For example, they can activate platelets by binding to platelet-specific receptor glycoprotein VI (GPVI) [71]. PS oligonucleotides have a complex immunostimulatory effect e.g., they induce interferone synthesis and may increase the lipopolysaccharide-caused TNF-α synthesis, probably by mimicking the structure of the polyanionic glycosaminoglycans in the ECM [72]. There are also sequence-dependent effects, e.g., the CpG motif (especially the purine-purine-CG-pyrimidine-pyrimidine sequence) can stimulate B cells [73] as the CG sequence is rare and even methylated in vertebrates, while common in prokaryotes [74]. Therefore, the Toll-like receptor 9 (TLR9) of the human immune system recognizes this sequence as a bacterial pattern [75]. PS oligonucleotides strongly bind to plasma proteins and after systemic absorption, the liver and the kidney are the main sites of accumulation. In case of intravitreal administration, no significant systemic absorption was observed. Their metabolism is not primarly mediated by the CYP-450 enzyme family (which enzymes play important role in the metabolism of most xenobiotics), but by nucleases, mainly 3′-exonucleases, which cleave nucleotides from the 3′-end of oligomers. The cellular uptake of PS oligos can also be explained by their interactions with cell membrane proteins [70][76]. The PS modification is part of the first and second generation ASOs. In N3′→P5′ phosphoramidate oligodeoxynucleotides, the 3′-oxygen of nucleotides is replaced by a nitrogen. This modification increases the stability of duplexes formed with DNA and RNA. In addition, these derivatives with homopyrimidine sequences are able to form triplexes with DNA or RNA [77][78]. Incorporation of an electronegative group (fluorine) in the 2′ position of N3′→P5′ phosphoramidate oligos increases [79], while an electron donating group (OMe) in the same position decreases the stability of duplexes [80], presumably by modifying the conformation of the furanose ring. The N-methyl derivatives (3′NMe→P5′ phosphoramidate) cannot hybridize to RNA or DNA [81].
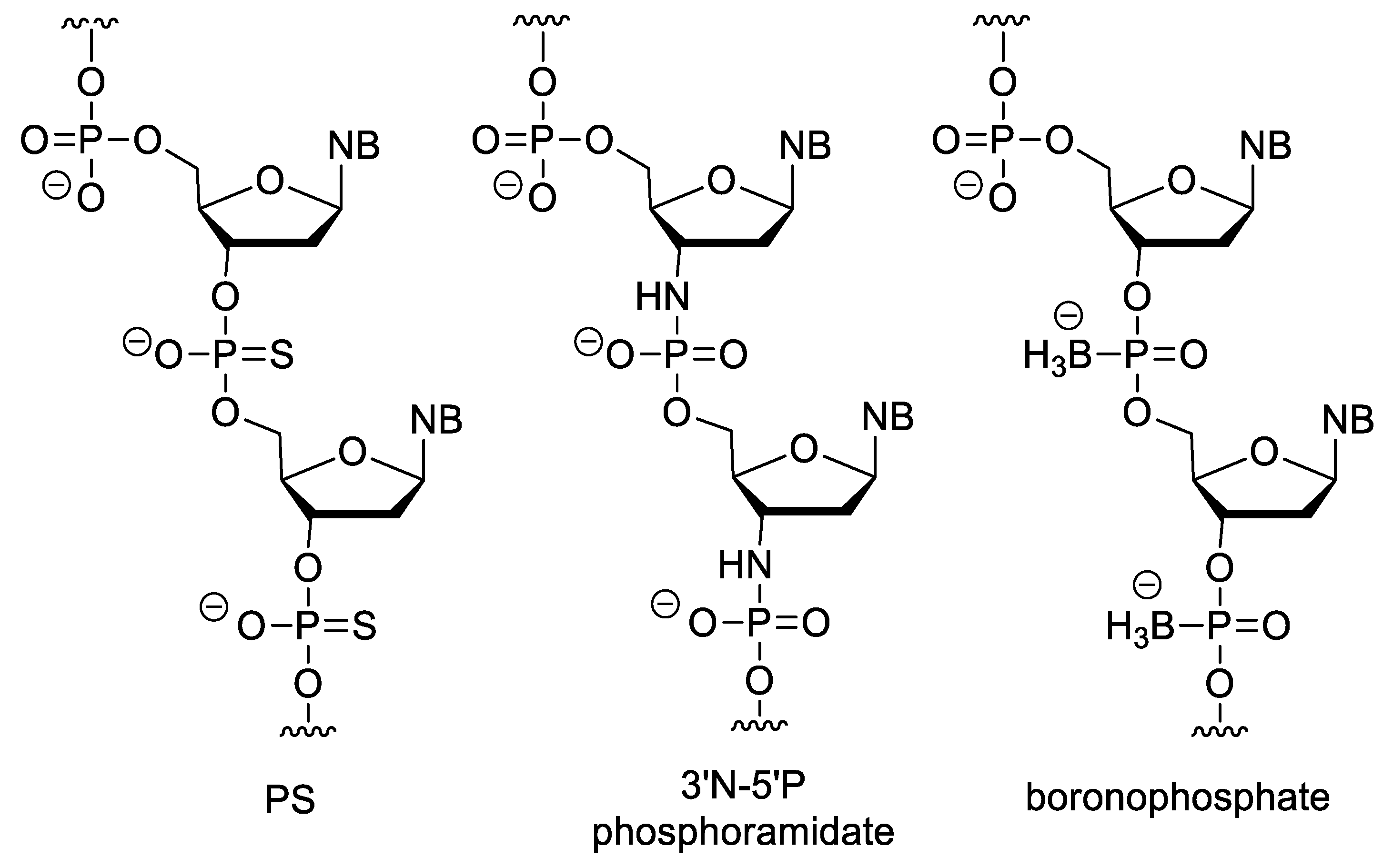
Figure 7. XNAs with negatively charged backbone.
In boronophosphate oligonucleotides, one of the non-bridging oxygen of the phosphate ester is replaced by a BH3 group. Because of their anionic backbone, they show good water solubility, but are more lipophilic than DNA. They can be synthesized similarly to oligonucleotide synthesis, but the oxidation with iodine has been replaced by boronation of the protected H-phosphonate derivative with a borane-amine complex [78][82][83].
It is very useful to change the polyanionic phosphate ester backbone to an uncharged one because in this case the electrostatic repulsion that occurs during hybridization between the two strands is eliminated, which can cause stronger binding. In addition, due to the lack of the electric repulsion between the backbone and the negatively charged cell membrane surface, the cellular uptake can also be improved this way. The above mentioned PMOs (see Figure 3) with their phosphorodiamidate internucleotide linkages are also electrostatically neutral.
In the alkylphosphonate oligonucleotides, one of the non-bridging oxygen of the phosphate group is replaced by an alkyl group. Among these, the methylphosphonates (MPO) are the most important (Figure 8). Their synthesis is simple by using methylphosphonoamidites in conventional oligonucleotide synthesis [84][85]. The MPOs are very resistant against nucleases. However, their affinity to natural nucleic acids is low, and they cannot activate RNAse H [78]. The incorporation of methylphosphonothioate units (MPS) into oligonucleotides slightly decreases the stability of duplexes, while significantly increasing the half-life of the oligomer [86][87][88][89][90]. Phosphotriester (PT) oligos such as methylphosphotriester (MPT) derivatives have better cellular uptake due to their neutral backbone. Inside the cell, PTs are enzymatically transformed into native oligonucleotides, so they can be used as prodrugs of siRNAs [91][92].
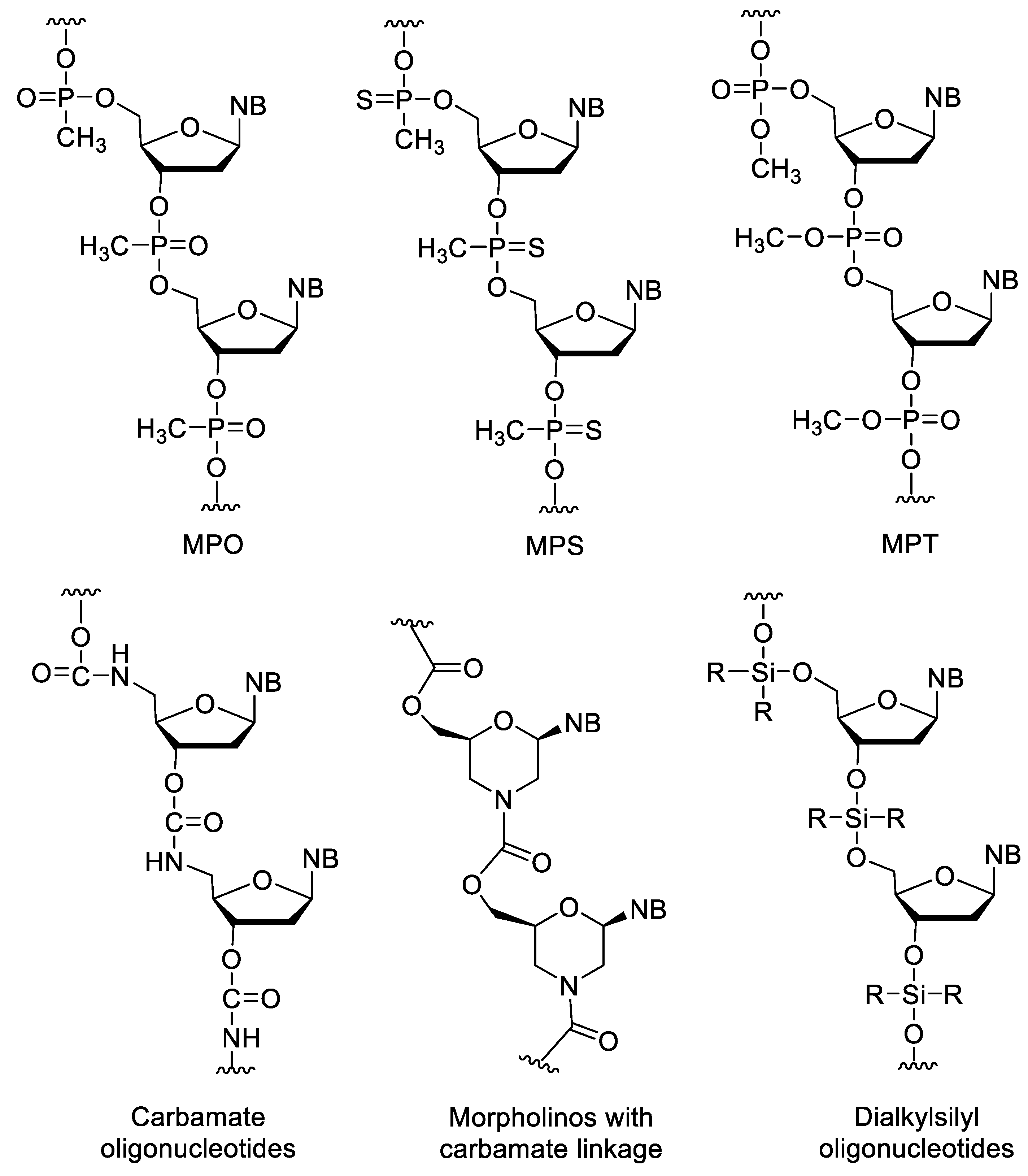
Figure 8. XNAs with neutral backbone.
Carbamate (CA) oligos are stable under acidic and alkaline conditions and are resistant to some enzymes [86][93]. There are also morpholinos with carbamate linkages [94]. Dialkylsilyl oligonucleotides are acid sensitive and have very low water solubility [95][96].
Peptide nucleic acids (PNAs) are very uniqe representatives of XNAs. PNA was developed in 1991 by Nielsen et al. [97]. In the original PNA, the sugar-phosphate backbone of the oligonucleotides is replaced by a poly-aminoethylglycine (Aeg), to which the nucleobases are attached through a methylenecarbonyl linker (Figure 9). This structure has several advantages, it is acyclic, achiral, electrostatically neutral, fully resistant to nucleases and highly resistant to peptidases. PNA are not sensitive to acidic or alkaline conditions. They bind excellently to DNA and RNA with high affinity and selectivity, and are also able to form triplexes with double-stranded DNA. The latter property enables PNA to anti-gene gene silencing. PNAs can be synthesized by simple peptide synthesis strategies. Their disadvantages are their limited water solubility, poor cellular uptake, and self-aggregation. The solubility decreases by increasing the length or the purine/pyrimidine ratio, but can be increased by incorporating ionic groups, and the cellular uptake can be improved by formulation or by using PNA/DNA chimeras [98]. In addition to the classic AegPNA, many other PNA derivatives have developed with a modified backbone, linker, or side chain [98][99].
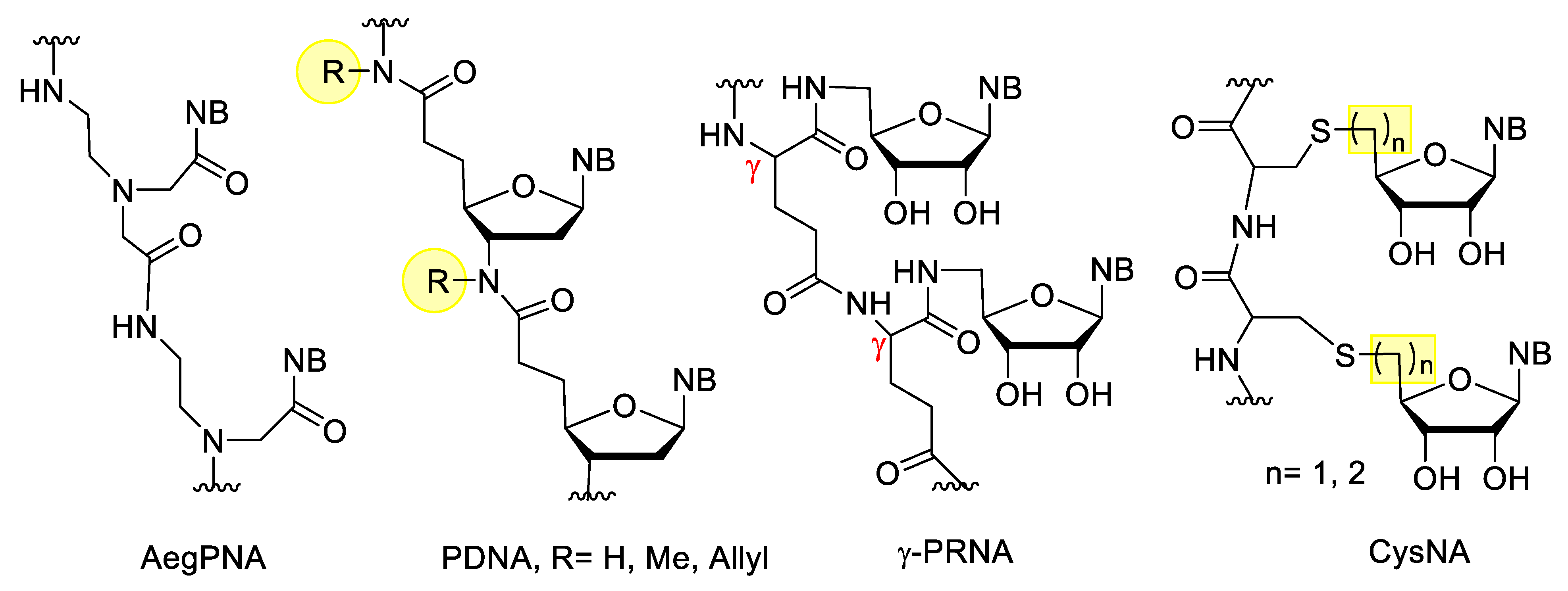
Figure 9. XNAs with peptide type backbones.
Besides peptide nucleic acids, there are other XNAs with peptide bonds which also contain the ribose component. The simplest examples include the peptide deoxyribonucleic acid (PDNA) derivatives, developed by Freier et al. [100]. Integration of these derivatives into oligonucleotides increased the metabolic stability, but decreased the binding affinity. In γ-peptide ribonucleic acid (γ-PRNA) the 5′-amino-5′deoxynucleosides are linked to the γ-peptide chain through an amide bond. These derivatives are more soluble in water than PNA and due to their free 2′,3′-OH groups, their hybridization can be controlled by borax, which can form borate with vicinal diols [101][102]. Cysteinyl nucleic acids (CysNA) contain an oligocysteine backbone, to which the nucleosides are attached through a thioether bond. These can be constructed from the monomers via conventional solid phase peptide synthesis (SPPS). The monomers can be synthesized by nucleophilic substitution of the 5′-activated nucleoside and cysteine, or by thiol-ene coupling reaction between cysteine and the unsaturated nucleoside derivative [103].
Besides the anionic and neutral NAs, nucleic acid analogues with positively charged backbones have also been developed. Their general advantages over anionic derivatives are their better cellular uptake, resistance to nucleases and higher binding affinity. Their theoretical common disadvantage may be their non-specific binding to proteins or to the polyanionic backbone of nucleic acids. However, in practice this is not a major problem, their sequence specificity is not decreased significantly and the XNA–protein interactions are a more serious problem for the anionic PS oligos [104][105]. The deoxyribonucleic guanidine oligomer (DNG) contains a guanidine bridge between the two nucleosides (Figure 10). DNG can form stable duplexes with complementary DNA, and poly-A or poly-T DNG can also form triplexes with the complementary DNA [104][105][106]. Relatives of DNG are the deoxyribonucleic methylthiourea (DNmt) oligonucleotides that contain an S-methylthiourea linker. The properties of DNmt are basically similar to the DNG. While in the case of anionic XNAs, higher ionic strength can stabilize duplexes by coating the negative charges of the backbone with cations, the stability of DNmt/DNA duplexes decreases at higher ionic concentrations [104][105][107].
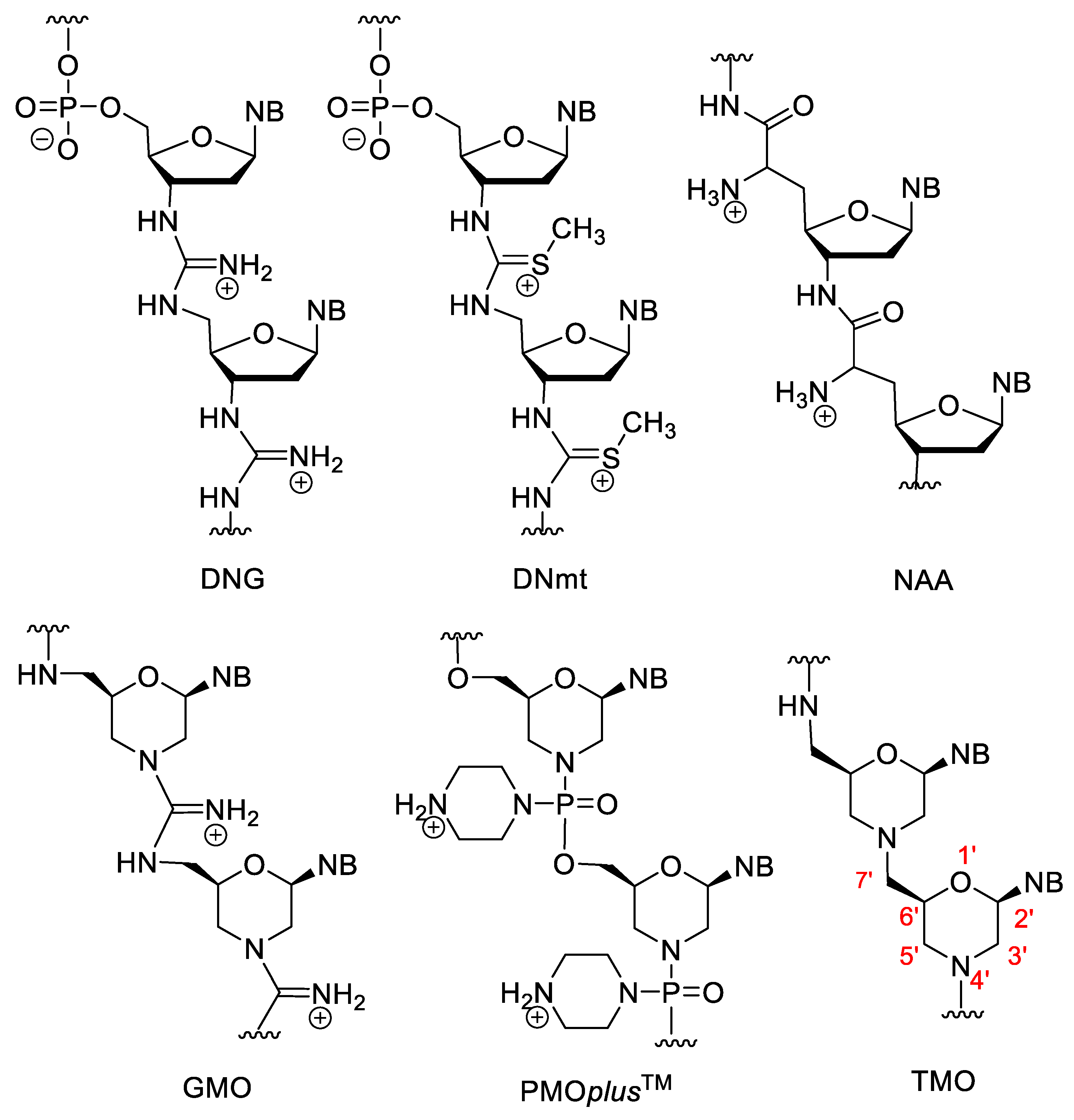
Figure 10. XNAs with positively charged backbone.
Nucleosyl amino acids (NAA) are similar to the amide derivatives mentioned above, but the backbone is susbtituted by primary amino groups, which impart a cationic character to the molecule. NAA has good hybridization strength, especially the (S)-isomer, however, it has low mismatch discrimination character [105][108]. By incorporation of NAA units into DNA, zwitterionic oligonucleotides were obtained [109].
There are various positively charged morpholino oligomers. Guanidine-linked morpholinos (GMOs) have better cellular uptake than electroneutral ones [110]. In PMOplusTM, the nitrogen of the phosphorodiamidate group is part of a piperazine ring [40][111]. PMOplusTM has been tested against various viruses, e.g., influenza virus [112]. In tightly linked morpholino oligomers (TMO), the 7′-C is directly connected to the nitrogen atom of the morpholino ring, creating a tertiary amino group, which is protonated in aqueous solution. These derivatives are synthesized by the double reductive amination-cyclocondensation reaction of nucleoside-derived secodialdehydes and 5′-amino-5′-deoxy nucleosides [113].
References
- Wan, W.B.; Seth, P.P. The Medicinal Chemistry of Therapeutic Oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667.
- Kumar, P.; Caruthers, M.H. DNA Analogues Modified at the Nonlinking Positions of Phosphorus. Acc. Chem. Res. 2020, 53, 2152–2166.
- Reese, C.B. Oligo- and poly-nucleotides: 50 years of chemical synthesis. Org. Biomol. Chem. 2005, 3, 3851–3868.
- Verma, S.; Eckstein, F. Modified Oligonucleotides: Synthesis and Strategy for Users. Annu. Rev. Biochem. 1998, 67, 99–134.
- Scremin, C.L.; Zhou, L.; Srinivasachar, K.; Beaucage, S.L. Stepwise Regeneration and Recovery of Deoxyribonucleoside Phosphoramidite Monomers during Solid-Phase Oligonucleotide Synthesis. J. Org. Chem. 1994, 59, 1963–1966.
- Flanagan, W.M.; Wolf, J.J.; Olson, P.; Grant, D.; Lin, K.; Wagner, R.W.; Matteucci, M.D. A cytosine analog that confers enhanced potency to antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1999, 96, 3513–3518.
- Chenna, V.; Rapireddy, S.; Sahu, B.; Ausin, C.; Pedroso, E.; Ly, D.H. A Simple Cytosine to G-Clamp Nucleobase Substitution Enables Chiral γ-PNAs to Invade Mixed-Sequence Double-Helical B-form DNA. ChemBioChem 2008, 9, 2388–2391.
- Holmes, S.C.; Arzumanov, A.A.; Gait, M.J. Steric inhibition of human immunodeficiency virus type-1 Tat-dependent trans-activation in vitro and in cells by oligonucleotides containing 2′-O-methyl G-clamp ribonucleoside analogues. Nucleic Acids Res. 2003, 31, 2759–2768.
- Wojciechowski, F.; Hudson, R.H.E. A Fmoc/Boc pseudoisocytosine monomer for peptide nucleic acid synthesis. Can. J. Chem. 2008, 86, 1026–1029.
- Neuer, P.; Monaci, P. New Fmoc Pseudoisocytosine Monomer for the Synthesis of Bis-PNA Molecule by Automated Solid-Phase Fmoc Chemistry. Bioconjugate Chem. 2002, 13, 676–678.
- Compagno, D.; Lampe, J.N.; Bourget, C.; Kutyavin, I.V.; Yurchenko, L.; Lukhtanov, E.A.; Gorn, V.V.; Gamper, H.B.; Toulmé, J. Antisense oligonucleotides containing modified bases inhibit in vitro translation of Leishmania amazonensis mRNAs by invading the mini-exon hairpin. J. Biol. Chem. 1999, 274, 8191–8198.
- Kutyavin, I.V.; Rhinehart, R.L.; Lukhtanov, E.A.; Gorn, V.V.; Meyer, R.B.; Gamper, H.B. Oligonucleotides Containing 2-Aminoadenine and 2-Thiothymine Act as Selectively Binding Complementary Agents. Biochemistry 1996, 35, 11170–11176.
- Patil, K.M.; Toh, D.K.; Yuan, Z.; Meng, Z.; Shu, Z.; Zhang, H.; Ong, A.A.L.; Krishna, M.S.; Lu, L.; Lu, Y.; et al. Incorporating uracil and 5-halouracils into short peptide nucleic acids for enhanced recognition of A-U pairs in dsRNAs. Nucleic Acids Res. 2018, 46, 7506–7521.
- Sági, J.; Szemző, A.; Ebinger, K.; Szabolcs, A.; Sági, G.; Ruff, É.; Ötvös, L. Base Modified oligodeoxynucleotides I. Effect of 5-Alkyl, 5-(1-Alkenyl), and 5-(1-Alkinyl) Substitution of the Pirimidines on Duplex Stability and Hydrophobicity. Tetrahedron Lett. 1993, 35, 2191–2194.
- Wlotzka, B.; Leva, S.; Eschgfäller, B.; Burmeister, J.; Kleinjung, F.; Kaduk, C.; Muhn, P.; Hess-Stumpf, H.; Klussmann, S. In vivo properties of an anti-GnRH Spiegelmer: An example of an oligonucleotide based therapeutic substance class. Proc. Natl. Acad. Sci. USA 2002, 13, 8898–8902.
- Vater, A.; Klussmann, S. Turning-mirror image oligonucleotides into drugs: The evolution of Spiegelmer therapeutics. Drug Discov. Today 2015, 20, 147–155.
- Latha, Y.S.; Yathrinda, N. Stereochemical Studies on Nucleic Acid Analogues I. Conformations of α-Nucleosides and α-Nucleotides: Interconversion of Sugar Puckers via O-4′exo. Bipolymers 1992, 32, 249–269.
- Boiziau, C.; Kurfurst, R.; Cazenave, C.; Roig, V.; Thoung, N.T.; Toulmé, J.J. Inhibition of translation initiation by antisense oligonucleotides via an RNase-H independent mechanism. Nucleic Acid Res. 1991, 19, 1113–1119.
- Frieden, M.; Christensen, S.M.; Mikkelsen, N.D.; Rosenbohm, C.; Thrue, C.A.; Westergaard, M.; Hansen, H.F.; Orum, H.; Koch, T. Expanding the design Horison of antisense oligonucleotides with alpha-L-LNA. Nucleic Acids Res. 2003, 31, 6365–6372.
- Eschenmoser, A.; Dobler, M. Warum Pentose und nicht Hexose-Nukleinsäuren? Teil I. Einleitung und Problemstellung, Konformationsanalyse fur Oligonucleotid-Ketten aus 2′, 3′-Dideoxyglucopyranosyl-Bausteinen (‘Homo-DNS’) sowie Betrachtungen zur Konformation von A-und B-DNS. Helv. Chim. Acta 1992, 75, 218–259.
- Bohringer, M.; Roth, H.; Hunziker, J.; Gobe, M.; Krishnan, R.; Giger, A.; Schweizer, B.; Schreiber, J.; Leumann, C.; Eschenmoser, A. Warum Pentose und nicht Hexose-Nukleinsäuren? Teil II. Oligonucleotide aus 2′,3′-Dideoxy-β-d-glucopyranosyl-Bausteinen (‘Homo-DNS’): Herstellung. Helv. Chim. Acta 1992, 75, 1416–1477.
- Hunziker, J.; Roth, H.; Böhringer, M.; Giger, A.; Diederischen, U.; Göbel, M.; Krishnan, R.; Jaun, B.; Leumann, C.; Eschenmoser, A. Warum Pentose und nicht Hexose-Nukleinsäuren? Teil III. Oligo(2′,3′-dideoxy-β-D-glucopyranosyl)nucleotide (‘Homo-DNS’) Paarungseigenschaften. Helv. Chim. Acta 1993, 76, 259–352.
- Egli, M.; Lubini, P.; Pallan, P.S. The long and winding road to the structure of homo-DNA. Chem. Soc. Rev. 2007, 36, 31–45.
- Egli, M.; Pallan, P.S.; Pattanayek, R.; Wilds, C.J.; Lubini, P.; Minsaov, G.; Dobler, M.; Leumann, C.J.; Eschenmoser, A. Crystal Structure of Homo-DNA and Nature’s Choice of Pentose over Hexose int he Genetic System. J. Am. Chem. Soc. 2006, 128, 10847–10856.
- Pitsch, S.; Krishnamurthy, R.; Bolli, M.; Wendehorn, S.; Holzner, A.; Minton, M.; Lesuenr, C.; Schlonvogt, I.; Jaun, B.; Eschenmoser, A. Pyranosyl-RNA (‘p-RNA’): Base-Pairing Selectivity and Potential to Replicate. Helv. Chim. Acta 1995, 78, 1621–1635.
- Beier, M.; Reck, F.; Wagner, T.; Krishnamurty, R.; Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure: Comparing Pentopyranosyl-(2′-4′) Oligonucleotides with RNA. Science 1999, 283, 699–703.
- Reck, F.; Wippo, H.; Kudick, R.; Bolli, M.; Ceulemans, G.; Krishnamurthy, R.; Eschenmoser, A. L-r-Lyxopyranosyl (4′-3′) Oligonucleotides: A Base-Pairing System Containing a Shortened Backbone. Org. Lett. 1999, 1, 1531–1534.
- Jungmann, O.; Wippo, H.; Stanek, M.; Huynh, H.K.; Krishnamurthy, R.; Eschenmoser, A. Promiscuous Watson−Crick Cross-Pairing within the Family of Pentopyranosyl (4′-2′) Oligonucleotides. Org. Lett. 1999, 1, 1527–1530.
- Schöning, K.; Scholz, P.; Guntha, S.; Wu, X.; Krishnamurthy, R.; Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure: The α-Threofuranosyl-(3′-2′) Oligonucleotide System. Science 2000, 290, 1347–1351.
- Noronha, A.M.; Wilds, C.J.; Lok, C.; Viazovkina, K.; Arion, D.; Parniak, M.A.; Damha, M.J. Synthesis and Biophysical Properties of Arabinonucleic Acids (ANA): Circular Dichroic Spectra, Melting Temperatures, and Ribonuclease H Susceptibility of ANA∙RNA Hybrid Duplexes. Biochemistry 2000, 39, 7050–7062.
- Wilds, C.J.; Damha, M.J. 2′-Deoxy-2′-fluoro-β-D-arabinonucleosides and oligonucleotides (2′F-ANA): Synthesis and physicochemical studies. Nucleic Acids Res. 2000, 28, 3625–3635.
- Maurinsh, Y.; Rosemeyer, H.; Esnouf, R.; Medvedovici, A.; Wang, J.; Ceulemans, G.; Lescrinier, E.; Hendrix, C.; Busson, R.; Sandra, P.; et al. Synthesis and Pairing Properties of Oligonucleotides Containing 3-Hydroxy-4-hydroxymethyl-1-cyclohexanyl Nucleosides. Chem. Eur. J. 1999, 5, 2139–2150.
- Wang, J.; Verbeure, B.; Luyten, I.; Lescrinier, E.; Froeyen, M.; Hendrix, C.; Rosemeyer, H.; Seela, F.; Aerschot, A.; Herdewijn, P. Cyclohexene Nucleic Acids (CeNA): Serum Stable Oligonucleotides that Activate RNase H and Increase Duplex Stability with Complementary RNA. J. Am. Chem. Soc. 2000, 122, 8595–8602.
- Verbeure, B.; Lescrinier, E.; Wang, J.; Herdewijn, P. RNAse H mediated cleavage of RNA by cyclohexene nucleic acid (CeNA). Nucleic Acid Res. 2001, 29, 4941–4947.
- D’Alonzo, D.; Froeyen, M.; Schepers, G.; Fabio, G.D.; Aerschot, A.; Hardewijn, P.; Palumbo, G.; Guaragna, A. 1′, 5′-Anhydro-L-ribo-hexitol Adenine Nucleic Acids (α-L-HNA-A): Synthesis and Chiral Selection Properties in the Mirror Image World. J. Org. Chem. 2015, 80, 5014–5022.
- Bouvere, B.D.; Kerremans, L.; Hendrix, C.; Winter, H.D.; Schepers, G.; Aerschot, A.; Herdewijn, P. Hexitol Nucleic Acids (HNA): Synthesis and Properties. Nucleosides Nucleotides 1997, 16, 973–976.
- Kang, H.; Fischer, M.H.; Xu, D.; Miyamoto, Y.J.; Marchand, A.; Aerschot, A.; Herdewijn, P.; Juliano, R.L. Inhibition of MDR1 gene expression by chimeric HNA antisense oligonucleotides. Nucleic Acid. Res. 2004, 32, 4411–4419.
- Fisher, M.; Abramov, M.; Aerschot, A.; Rozenski, J.; Dixit, V.; Juliano, R.L.; Herdewijn, P. Biological effects of hexitol and altritol-modified siRNAs targeting B-Raf. Eur. J. Pharmacol. 2009, 606, 38–44.
- Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195.
- Nan, Y.; Zhang, Y. Antisense Phosphorodiamidate Morpholino Oligomers as Novel Antiviral Compounds. Front. Microbiol. 2018, 9, 750.
- Corey, D.R.; Abrams, J.M. Morpholino antisense oligonucleotides: Tools for investigating vertebrate development. Genome Biol. 2001, 2, 1015.1.
- Nasevicius, A.; Ekker, S.C. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000, 26, 216–220.
- Heasman, J.; Kofron, M.; Wylie, C. β-Catenin Signaling Activity Dissected in the Early Xenopus Embryo: A Novel Antisense Approach. Dev. Biol. 2000, 222, 124–134.
- Howard, E.W.; Newman, L.A.; Oleksyn, D.W.; Angerer, R.C.; Angerer, L.M. SpKrl: A direct target of β-catenin regulation required for endoderm differentiation in sea urchin embryos. Development 2001, 128, 365–375.
- Bolli, M.; Litten, J.C.; Schütz, R.; Leumann, C.J. Bicyclo-DNA: A Hoogsteen-selective pairing system. Chem. Biol. 1996, 3, 197–206.
- Tarköy, M.; Bolli, M.; Leumann, C. Nucleic-Acid Analogues with Restricted Conformational Flexibility in the Sugar-Phosphate Backbone (‘Bicyclo-DNA’) (Part 3) Synthesis, Pairing Properties, and Calorimetric Determination of Duplex and Triplex Stability of Decanucleotides from adenine and -thymine. Helv. Chim. Acta 1994, 77, 716–744.
- Steffens, R.; Leumann, C.J. Tricyclo-DNA: A Phosphodiester-Backbone Based DNA Analog Exhibiting Strong Complementary Base-Pairing Properties. J. Am. Chem. Soc. 1997, 119, 11548–11549.
- Renneberg, D.; Bouliong, E.; Reber, U.; Schümperli, D.; Leumann, C.J. Antisense properties of tricyclo-DNA. Nucleic Acid Res. 2002, 30, 2751–2757.
- Aupy, P.; Echevarría, L.; Relizani, K.; Goyenvalle, A. The Use of Tricyclo-DNA Oligomers for the Treatment of Genetic Disorders. Biomedicines 2018, 6, 2.
- Renneberg, D.; Leumann, C.J. Watson-Crick Base-Pairing Properties of Tricyclo-DNA. J. Am. Chem. Soc. 2002, 124, 5993–6002.
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the Adenine, Cytosine, Guanine, 5-Methylcytosine, Thymine and Uracil Bicyclonucleoside Monomers, Oligomerisation, and Unprecedented Nucleic Acid Recognition. Tetrahedron 1998, 54, 3607–3630.
- Kaur, H.; Babu, B.R.; Maiti, S. Perspectives on Chemistry and Therapeutic Applications of Locked Nucleic Acid (LNA). Chem. Rev. 2007, 107, 4672–4697.
- Langkjaer, N.; Pasternak, A.; Wengel, J. UNA (unlocked nucleic acid): A flexible RNA mimic that allows engineering of nucleic acid duplex stability. Bioorg. Med. Chem. 2009, 17, 5420–5425.
- Campbell, M.A.; Wengel, J. Locked vs. unlocked nucleic acids (LNA vs. UNA): Contrasting structures work towards common therapeutic goals. Chem. Soc. Rev. 2011, 40, 5680–5689.
- Merle, Y.; Bonneil, E.; Merle, L.; Sági, J.; Szemző, A. Acyclic oligonucleotide analogues. Int. J. Biol. Macromol. 1995, 17, 239–246.
- Heuberger, B.D.; Switzer, C. A Pre-RNA Candidate Revisited: Both Enantiomers of Flexible Nucleoside Triphosphates are DNA Polymerase Substrates. J. Am. Chem. Soc. 2008, 130, 412–413.
- Joyce, G.F.; Schwartz, A.W.; Miller, S.L.; Orgel, L.E. The case for an ancestral genetic system involving simple analogues of the nucleotides. Proc. Natl. Acad. Sci. USA 1987, 84, 4398–4402.
- Schlegel, M.K.; Peritz, A.E.; Kittigowittana, K.; Zhang, L.; Meggers, E. Duplex Formation of the Simplified Nucleic Acid GNA. ChemBioChem 2007, 8, 927–932.
- Schlegel, M.K.; Xie, X.; Zhang, L.; Meggers, E. Insight into the High Duplex Stability of the Simplified Nucleic Acid GNA. Angew. Chem. Int. Ed. 2009, 48, 960–963.
- Asanuma, H.; Toda, T.; Murayama, K.; Liang, X.; Kashida, H. Unexpectedly Stable Artificial Duplex from Flexible Acyclic Threoninol. J. Am. Chem. Soc. 2010, 132, 14702–14703.
- Murayama, K.; Kashida, H.; Asanuma, H. Acyclic L-threoninol nucleic acid (L-aTNA) with suitable structural rigidity cross-pairs with DNA and RNA. Chem. Commun. 2015, 51, 6500–6503.
- Kashida, H.; Murayama, K.; Asanuma, H. Acyclic artificial nucleic acids with phosphodiester bonds exhibit unique functions. Polym. J. 2016, 48, 781–786.
- Kashida, H.; Murayama, K.; Toda, T.; Asanuma, H. Control of the Chirality and Helicity of Oligomers of Serinol Nucleic Acid (SNA) by Sequence Design. Angew. Chem. Int. Ed. 2011, 50, 1285–1288.
- Freier, S.M.; Altmann, K. The ups and downs of nucleic acid duplex stability: Structure–stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acid Res. 1997, 25, 4429–4443.
- Baker, B.F.; Lot, S.S.; Condon, T.P.; Cheng-Flournoy, S.; Lesnik, E.A.; Sasmor, H.M.; Bennett, C.F. 2*-O-(2-Methoxy)ethyl-modified Anti-intercellular Adhesion Molecule 1 (ICAM-1) Oligonucleotides Selectively Increase the ICAM-1 mRNA Level and Inhibit Formation of the ICAM-1 Translation Initiation Complex in Human Umbilical Vein Endothelial Cells. J. Biol. Chem. 1997, 272, 11994–12000.
- Lind, K.E.; Mohan, V.; Manoharan, M.; Ferguson, D.M. Structural characteristics of 2′-O-(2-methoxyethyl)-modified nucleic acids from molecular dynamics simulations. Nucleic Acid Res. 1998, 26, 3694–3699.
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acid Res. 2020, 48, 2853–2865.
- Srinivasan, S.K.; Iversen, P. Review of In Vivo Pharmacokinetics and Toxicology of Phosphorothioate Oligonucleotides. J. Clin. Lab. Anal. 1995, 9, 129–137.
- Eckstein, F. Phosphorothioates, Essential Components of Therapeutic Oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387.
- Levin, A.A. A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys. Acta 1999, 1489, 69–84.
- Flierl, U.; Nero, T.L.; Lim, B.; Arthur, J.F.; Yao, Y.; Jung, S.M.; Gitz, E.; Pollitt, A.Y.; Zaldivia, M.T.K.; Jandrot-Perrus, M.; et al. Phosphorothioate backbone modifications of nucleotide-based drugs are potent platelet activators. J. Exp. Med. 2015, 212, 129–137.
- Hartmann, G.; Krug, A.; Waller-Fontaine, K.; Endres, S. Oligodeoxynucleotides Enhance Lipopolysaccharide- Stimulated Synthesis of Tumor Necrosis Factor: Dependence on Phosphorothioate Modification and Reversal by Heparin. Mol. Med. 1996, 2, 429–438.
- Jahrsdörfer, B.; Jox, R.; Mühlenhoff, L.; Tschoep, K.; Krug, A.; Rothenfusser, S.; Meinhardt, G.; Emmerich, B.; Endres, S.; Hartmann, G. Modulation of malignant B cell activation and apoptosis by bcl-2 antisense ODN and immunostimulatory CpG ODN. J. Leukoc. Biol. 2002, 72, 83–92.
- Karlin, S.; Ladunga, I.; Blaisdell, B.E. Heterogeneity of genomes: Measures and values. Proc. Natl. Acad. Sci. USA 1994, 91, 12837–12841.
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial RNA. Nature 2000, 408, 740–745.
- Crooke, S.T.; Vickers, T.A.; Liang, X. Phosphorothioate modified oligonucleotide–protein interactions. Nucleic Acid Res. 2020, 48, 5235–5253.
- Gryaznov, S.; Chen, J. ligodeoxyribonucleotide N3‘4P5’ Phosphoramidates: Synthesis and Hybridization Properties. J. Am. Chem. Soc. 1994, 116, 3143–3144.
- Micklefield, J. Backbone Modification of Nucleic Acids: Synthesis, Structure and Therapeutic Applications. Curr. Med. Chem. 2001, 8, 1157–1179.
- Schultz, R.G.; Gryaznov, S.M. Oligo-2′-fluoro-2′-deoxynucleotide N3′→P5′ phosphoramidates: Synthesis and properties. Nucleic Acid Res. 1996, 24, 2966–2973.
- Tereshko, V.; Gryaznov, S.; Egli, M. Consequences of Replacing the DNA 3′-Oxygen by an Amino Group: High-Resolution Crystal Structure of a Fully Modified N3′→P5′ Phosphoramidate DNA Dodecamer Duplex. J. Am. Chem. Soc. 1998, 120, 269–283.
- Gryaznov, S.M.; Lloyd, D.H.; Chen, J.; Schultz, R.G.; DeDionsio, L.A.; Ratmeyer, L.; Wilson, W.D. Oligonucleotide N3′→P5′ phosphoramidates. Proc. Natl. Acad. Sci. USA 1995, 92, 5798–5802.
- Sood, A.; Shaw, B.R.; Spielvogel, B.F. Boron-Containing Nucleic Acids. 2′ Synthesis of Oligodeoxynucleoside Boranophosphates. J. Am. Chem. Soc. 1990, 112, 9000–9001.
- Sergueev, D.S.; Shaw, B.R. H-Phosphonate Approach for Solid-Phase Synthesis of Oligodeoxyribonucleoside Boranophosphates and Their Characterization. J. Am. Chem. Soc. 1998, 120, 9417–9427.
- Reynolds, M.A.; Hogrefe, R.I.; Jaeger, J.A.; Schwartz, D.A.; Riley, T.A.; Marvin, W.B.; Daily, W.J.; Vaghefi, M.M.; Beck, T.A.; Knowles, S.K.; et al. Synthesis and thermodynamics of oligonucleotides containing chirally pure RP methylphosphonate linkages. Nucleic Acid Res. 1996, 24, 4584–4591.
- Flür, S.; Micura, R. Chemical synthesis of RNA with site-specific methylphosphonate modifications. Methods 2016, 107, 79–88.
- Clavé, G.; Reverte, M.; Vasseur, J.; Smietana, M. Modified internucleoside linkages for nuclease-resistant oligonucleotides. RSC Chem. Biol. 2021, 2, 94–150.
- Brill, W.K.D.; Caruthers, M.H. Synthesis of nucleoside methylphosphonothioates. Tetrahedron Lett. 1987, 28, 3205–3208.
- Pasmapriya, A.A.; Agrawal, S. Synthesis of Oligodeoxynucleoside methylphosphonothioates. Bioorg. Med. Chem. Lett. 1993, 3, 761–764.
- Wozniak, L.A.; Bukowiecka-Matusiak, M.; Gora, M.; Stec, W.J. One-Pot Synthesis of Dinucleoisde (3′,5′)-Methylphosphonothioates and their Seleno Congeners via the Phosphonotriazolidite Approach. Synlett 2006, 9, 1331–1334.
- Wozniak, L.A.; Gora, M.; Stec, W.J. Chemoselective Activation of Nucleoside 3′-O-Methylphosphonothioates with 1,3,5-Triazinyl Morpholinium Salts. J. Org. Chem. 2007, 72, 8584–8587.
- Hayakawa, Y.; Hirose, M.; Hayakawa, M.; Noyori, R. General Synthesis and Binding Affinity of Position-Selective Phosphonodiester- and Phosphotriester-Incorporated Oligodeoxyribonucleotides. J. Org. Chem. 1995, 60, 925–930.
- Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; Berg, A.; Hagopian, J.C.; Springer, A.D.; Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol. 2014, 32, 1256–1263.
- Mungall, W.S.; Kaiser, J.K. Carbamate Analogues of Oligonucleotides. J. Org. Chem. 1977, 42, 703–706.
- Stirchak, E.P.; Summerton, J.E.; Weller, D.D. Uncharged stereoregular nucleic acid analogs: 2. Morpholino nucleoside oligomers with carbamate internucleoside linkages. Nucleic Acid Res. 1989, 17, 6129–6141.
- Seliger, H.; Feger, G. Oligonucleotide Analogues with Dialkyl Silyl Internucleoside Linkages. Nucleosides Nucleotides 1987, 6, 483–484.
- Saha, A.K.; Sardaro, M.; Waychunas, C.; Delecki, D.; Rustny, R.; Cavanaugh, P.; Yawman, A.; Upson, D.A.; Kruse, L.I.; Kutny, R. Diisopropylsilyl-linked oligonucleotide analogs: Solid-phase synthesis and physicochemical properties. J. Org. Chem. 1993, 58, 7827–7831.
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a ThymineSubstituted Polyamide. Science 1991, 254, 1497–1500.
- Uhlmann, E.; Peyman, A.; Breipohl, G.; Will, D.W. PNA: Synthetic Polyamide Nucleic Acids with Unusual Binding Properties. Angew. Chem. Int. Ed. 1998, 37, 2796–2823.
- Das, A.; Pradhan, B. Evolution of Peptide Nucleic Acid with modifications of its backbone and application in Biotechnology. Chem. Biol. Drug Des. 2020, 97, 865–892.
- Lebreton, J.; Mesmaeker, A.; Waldner, A.; Fritsch, V.; Wolf, R.M.; Freie, S.M. Synthesis of Thymidine Dimer Derivatives Containing an Amide Linkage and their Incorporation into Oligodeoxyribonucleotides. Tetrahedron Lett. 1993, 34, 6383–6386.
- Wada, T.; Minamimoto, N.; Inaki, Y.; Inoue, Y. Peptide Ribonucleic Acids (PRNA). 2. A Novel Strategy for Active Control of DNA Recognition through Borate Ester Formation. J. Am. Chem. Soc. 2000, 122, 6900–6910.
- Sato, H.; Hashimoto, Y.; Wada, T.; Inoue, Y. Solid-phase synthesis of peptide ribonucleic acids (PRNA). Tetrahedron 2003, 59, 7871–7878.
- Bege, M.; Bereczki, I.; Molnár, D.J.; Kicsák, M.; Pénzes-Daku, K.; Bereczky, Z.; Ferenc, G.; Kovács, L.; Herczegh, P.; Borbás, A. Synthesis and oligomerization of cysteinyl nucleosides. Org. Biomol. Chem. 2020, 18, 8161–8178.
- Jain, M.L.; Bruice, P.Y.; Szabó, I.E.; Bruice, T.C. Incorporation of Positively Charged Linkages into DNA and RNA Backbones: A Novel Strategy for Antigene and Antisense Agents. Chem. Rev. 2012, 112, 1284–1309.
- Meng, M.; Ducho, C. Oligonucleotide analogues with cationic backbone linkages. Beilstein J. Org. Chem. 2018, 14, 1293–1308.
- Blaskó, A.; Dempcy, R.O.; Minyat, E.E.; Bruice, T.C. Association of Short-Strand DNA Oligomers with Guanidinium-Linked Nucleosides. A Kinetic and Thermodynamic Study. J. Am. Chem. Soc. 1996, 118, 7892–7899.
- Arya, D.P.; Bruice, T.C. Triple-helix formation of DNA oligomers with methylthiourealinked nucleosides (DNmt): A kinetic and thermodynamic analysis. Proc. Natl. Acad. Sci. USA 1999, 96, 4384–4389.
- Schmidtgall, B.; Kuepper, A.; Meng, M.; Grossmann, T.N.; Ducho, C. Oligonucleotides with Cationic Backbone and Their Hybridization with DNA: Interplay of Base Pairing and Electrostatic Attraction. Chem. Eur. J. 2017, 24, 1544–1553.
- Schmidtgall, B.; Spork, A.P.; Wachowius, F.; Höbartner, C.; Ducho, C. Synthesis and properties of DNA oligonucleotides with a zwitterionic backbone structure. Chem. Commun. 2014, 50, 13742–13745.
- Pattanayak, S.; Khatra, H.; Saha, S.; Sinha, S. A cationic morpholino antisense oligomer conjugate: Synthesis, cellular uptake and inhibition of Gli1 in the hedgehog signalling pathway. RSC Adv. 2014, 4, 1951–1954.
- Warren, T.K.; Shurtleff, A.C.; Bavari, S. Advanced morpholino oligomers: A novel approach to antiviral therapy. Antivir. Res. 2012, 94, 80–88.
- Meng, M.; Zhang, J.; Liu, A.; Reuschel, S.; Sazani, P.; Wong, M. Quantitative determination of AVI-7100 (Radavirsen), a phosphorodiamidate morpholino oligomer (PMOplus®), in human plasma using LC–MS/MS. Bioanalysis 2017, 9, 827–839.
- Debreczeni, N.; Bege, M.; Herczeg, M.; Bereczki, I.; Batta, G.; Herczegh, P.; Borbás, A. Tightly linked morpholino-nucleoside chimeras: New, compact cationic oligonucleotide analogues. Org. Biomol. Chem. 2021, 19, 8711–8721.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.4K
Revisions:
2 times
(View History)
Update Date:
05 Aug 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No