Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Chao Yang | -- | 2655 | 2022-08-03 14:00:00 | | | |
| 2 | Sirius Huang | + 1 word(s) | 2656 | 2022-08-04 05:21:14 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yang, C.; Chen, E.A.; Zhang, Y. Structure-Based Virtual Screening. Encyclopedia. Available online: https://encyclopedia.pub/entry/25812 (accessed on 21 July 2026).
Yang C, Chen EA, Zhang Y. Structure-Based Virtual Screening. Encyclopedia. Available at: https://encyclopedia.pub/entry/25812. Accessed July 21, 2026.
Yang, Chao, Eric Anthony Chen, Yingkai Zhang. "Structure-Based Virtual Screening" Encyclopedia, https://encyclopedia.pub/entry/25812 (accessed July 21, 2026).
Yang, C., Chen, E.A., & Zhang, Y. (2022, August 03). Structure-Based Virtual Screening. In Encyclopedia. https://encyclopedia.pub/entry/25812
Yang, Chao, et al. "Structure-Based Virtual Screening." Encyclopedia. Web. 03 August, 2022.
Copy Citation
Molecular docking plays a significant role in early-stage drug discovery, from structure-based virtual screening (VS) to hit-to-lead optimization. VS is a computational approach used to identify chemical structures that are predicted to have particular properties. In drug discovery, it involves computationally searching large libraries of chemical structures to identify those structures that are most likely to bind to a target protein.
molecular docking
virtual screening
machine learning
protein-ligand scoring function
1. Introduction
Structure-based virtual screening (VS), also known as target-based VS, attempts to predict the best interaction of a ligand against a target protein to form a complex and employs scoring functions to estimate the binding affinity of the protein–ligand complex [1]. As a result, all the ligands are ranked according to their binding scores to the target, and the high-scoring ligands are selected for experimental measurement. In recent decades, advances in VS have been made in the following:
- There have been developments in structure-based VS approaches, including improvements in sampling and scoring methods, that have resulted in significant improvements in docking, scoring and screening performances [2].
- Developments in GPU processing speeds and cloud computing have dramatically increased computational power. Researchers are now able to computationally process vast numbers of compounds in the drug-like chemical space.
- Advancements in structural biology (such as X-ray, nuclear magnetic resonance (NMR) and cryo-EM) and computational protein structure prediction (such as AlphaFold2 and RoseTTAFold) [3][4][5][6] have allowed access to many more 3D structures.
- The number of compounds that are commercially available or can be readily synthesized has grown dramatically in recent years. For example, as of March 2021, the WuXi GalaXi and Enamine REAL Space collections contain 2.1 billion and 17 billion compounds, respectively [7]. In June 2022, the WuXi GalaXI and Enamine REAL Space collections have grown up to 4.4 billion and 22.7 billion compounds, respectively.
The convergence of these breakthroughs has positioned structure-based VS to be a promising direction for the discovery of novel small molecule medicine. With the appropriate computing infrastructure, it becomes practical to virtually screen ultra-large compound library (synthesized or purchasable) to find virtual hit compounds, some of which (usually up to 100 compounds) can be experimentally tested.
2. Molecular Docking Protocol
Molecular docking methods predict receptor–ligand interactions at an atomic level and are widely utilized in structure-based VS. The docking process samples the optimal conformation based on the complementarity between the receptor and the ligand. Figure 1A shows the initially proposed “lock-and-key model”, which refers to the rigid docking of receptor and ligand to find the correct orientation for the “key” to open the “lock”. This model emphasizes the importance of geometric complementarity [8]. However, the real binding process is very flexible whereby the receptor and ligand changes their conformation to complement each other well. As shown in Figure 1B, the induced fit model considers structural flexibility and selects the lowest-energy bound state. Currently, major limitations of docking methods include a restricted sampling of both ligand and receptor conformations in pose prediction, as well as the previously discussed limited accuracy of scoring functions in affinity prediction.
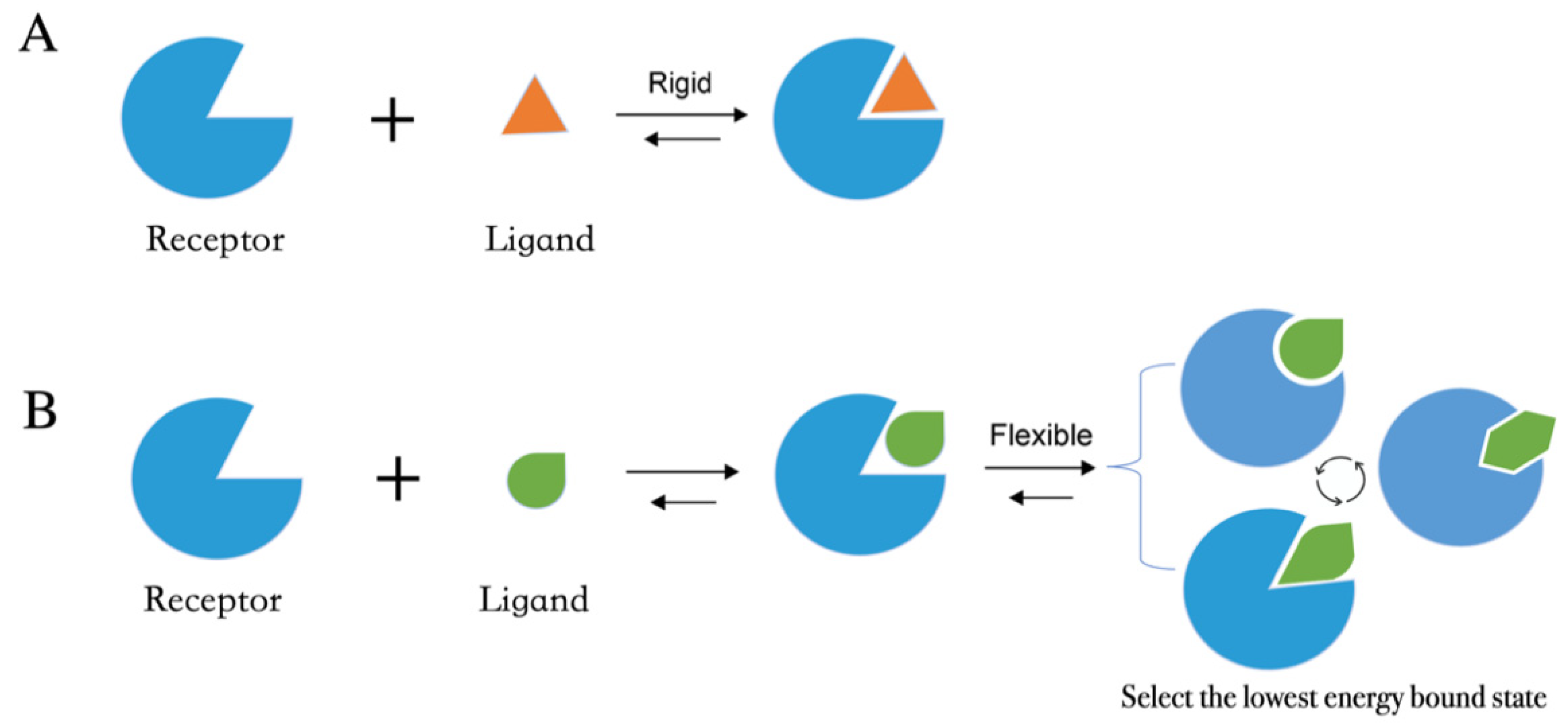
Figure 1. Two models of molecular docking. (A) A lock-and-key model. (B) Induced fit model.
The methods that improve sampling of ligand conformations can be defined as (i) incremental ligand construction, (ii) multiple conformers generation for docking and (iii) stochastic sampling [9]. In the first approach, the ligands are partitioned into small fragments that are individually docked into the receptor pocket according to the geometric fit. Docked fragments are then incrementally assembled to form an entire ligand within the binding pocket [10]. In the second approach, multiple low-energy conformations of the ligand are generated at first, and then individually docked against the receptor pocket [11].
The third and widely used strategy to account for ligand flexibility are stochastic methods, such as Monte Carlo (MC) or genetic algorithm (GA). MC algorithm, also known as simulated annealing, simulates docking by randomly generating minor changes in the position, orientation or conformation to generate new poses that are accepted or rejected based on the Metropolis acceptance algorithm [12]. The modeling begins at a high temperature such that there is a high probability of accepting the next conformation sampled. Then, the temperature is progressively decreased to reduce the conformational freedom of the system and to capture the receptor–ligand complex in a low energy state. GA employs a different approach inspired by Darwin’s theory of evolution [13]. The ligand begins as a random population of position, orientation and conformational states modeled as a set of chromosomes. Then, random crossovers and mutations are performed to produce another set of conformations. The conformation with the lowest binding energies with the receptor is accepted and then used to produce a new generation. This cycle is iteratively repeated until the local energy minimum of the receptor–ligand complex has been reached.
Many proteins possess varying degrees of flexibility, which can range from a slight perturbation of the ligand binding pocket to a complete reconstitution of the pocket. Therefore, an inadequate sampling of protein flexibility can result in an increase of both false positives and false negatives in VS experiments. Several approaches have been developed to tackle the issue of protein flexibility in recent years [14]. One common approach, named “ensemble docking”, is to utilize multiple receptor conformations in docking runs and to select the best-scoring conformation for further investigation [15][16][17]. The receptor conformations are commonly obtained from different X-ray and NMR structures or by sampling structures from molecular dynamics (MD) simulations. For instance, Abagyan and co-workers have investigated strategies for the selection of experimental protein conformations for VS and have found that the use of ensemble conformations of receptors co-crystallized with larger ligands provided the best results [18][19]. However, it has been noted that the use of excessively large numbers of receptor conformers in ensemble docking can lead to an increased number of false positive samples and linearly increased computational costs [14][20]. To alleviate some of these performance issues, ML techniques can be employed to help classify active and inactive compounds following ensemble docking [21]. Chandak and co-workers have tested multiple supervised ML methods trained on the DUD-E database to learn the relationship of a compound’s predicted binding affinities to the classification task.
An alternative approach to account for protein flexibility is to employ “soft docking”, where the interactions between the protein amino acid sidechains and the ligand is iteratively changed to allow partial clashing between the atoms of the protein and ligand [22]. For example, Ravindranath and co-workers have proposed a soft docking program, AutoDockFR [23], which simulates sidechain flexibility by sampling a large number of explicitly specified receptor sidechains and searching for energetically favorable binding poses for a given ligand. AutoDockFR optimizes protein–ligand interactions using the AutoDock4 force field and using a GA method combined with a Solis-Wets local search. This soft docking approach has achieved better binding pose prediction compared to rigid protein docking protocols but has also been associated with an increased number of false positive hits in structure-based VS [24].
3. Workflow in Virtual Screening
Structure-based VS relies on docking of large collections of compounds into the binding pocket of target protein, and then evaluating whether the protein–ligand contacts will drive binding. As shown in Figure 2, the general VS workflow can be as follows:
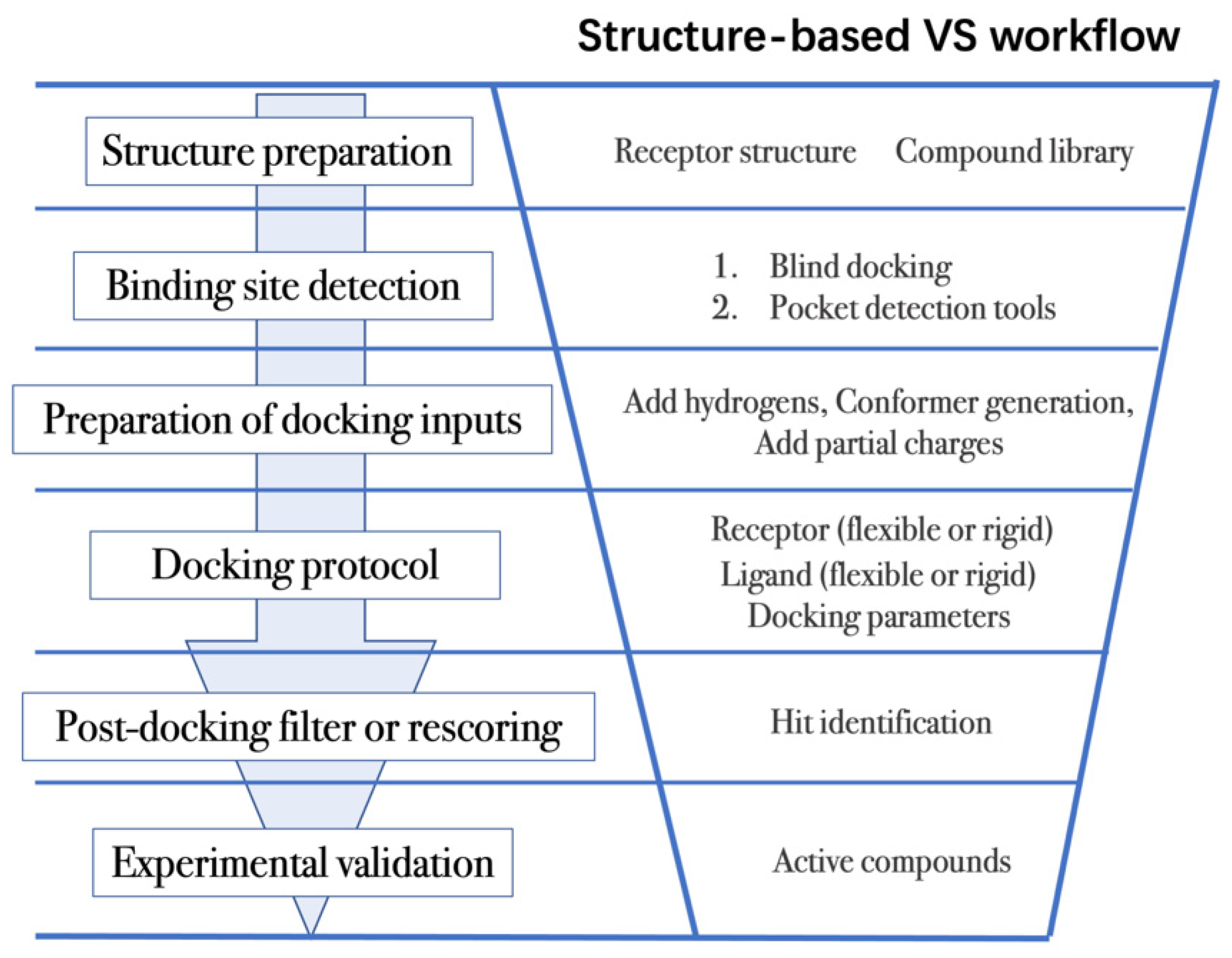
Figure 2. General scheme of a VS workflow.
- (1)
-
The first step is to obtain the 3D structures of a given target as well as the compound library. Experimental determined structures can be readily retrieved from the Protein Data Bank (PDB) [25], in which more than 120,000 unique protein structures have been determined through an enormous experimental effort. However, this represents a small fraction of the billions of known protein sequences whereby the 3D structure of a novel target is usually not available. In order to overcome this limitation, traditional computational prediction methods (such as homolog modelling and ab initio modelling) [26][27], as well as the recently developed DL methods (such as AlphaFold2 and RoseTTAFold) [3][5] can be employed to obtain the 3D structures of target proteins. In addition, the compound library or chemical space used in VS is also vital for hit identification.
As discussed above there is a growing number of options to dock to. It is important to note that the selection of which structure to dock to is not trivial. Docking results will differ depending on the conformation, apo/holo status, and quality of structure. One method, screening performance index, can be used to select good structures to use in prospective VS [28]. This index consists of five calculated terms that describe the docking performance of a set of structures on a set of known active compounds. Their testing has generally indicated that co-crystal structures with large ligands bound score well on the index and can be picked for prospective studies. These methods are limited because they require labeled datasets which may not be available for novel targets.
Compound libraries of approved drugs, natural products, already synthesized or purchasable compounds/fragments are commonly used in VS campaigns [9][29][30]. The well-known ZINC database contains over 750 million purchasable compounds, including over 230 million compounds in ready-to-dock 3D formats [31][32]. Recently, Jiankun and coworkers performed docking-based VS using a ultra-large compound library (more than 100 million compounds from ZINC make-on-demand compounds) to discover inhibitors targeting AmpC β-lactamase and D4 dopamine receptor [29]. Other databases, such as DrugBank [33][34][35] and Human Metabolome Database (HMDB) [36][37][38][39] are used to repurpose the approved drugs or human metabolites to the novel targets.
- (2)
-
The next step is to detect the binding site. Typically, the binding pocket on which to focus the docking calculations is known. For example, the binding site is chosen based on the information of co-crystallized ligand/substrate binding site, such as ATP binding site or protein–protein interactions (PPI) interface. However, when the binding site information is missing or a novel binding pocket needs to be explored, there are two commonly employed approaches, “blind docking” simulation [40][41] and pocket prediction algorithms. The first approach uses docking methods to search over the entire target structure to find a favorable ligand binding site, but it has a high computational cost in sampling. For the second approach, several available software can be employed to detect binding pockets, including AlphaSpace [42][43], FTMap [44], MDpocket [45], Fpocket [46], SiteMap [47] etc. These methods detect concave pockets on the protein surface by characterizing the spatial composition of amino acids or using the chemical probe to find favorable hot spots. Since drug resistance can arise for the orthosteric site of target proteins, these methods can be used to identify additional binding pockets that can be exploited for the design of novel inhibitors, such as allosteric or cryptic pockets [48][49].
- (3)
-
Once the binding site is determined it is important to carefully prepare docking input files to achieve successful VS. The preparation of protein structures starts from the assignment of protonation states for the amino acids, which can be done using software including PROPKA [50], H++ [51], and SPORES [52]. Then hydrogen atoms and partial charges are assigned. A popular software for this task is PDB2PQR [53][54]. In addition, the consideration of water molecules and metal ions can be crucial in certain target structures. Explicit water molecules mediating protein–ligand interactions should be analyzed and can be used to identify water-mediated interactions and avoid incorrect binding poses [55][56][57]. It is also important to consider coordination interactions between metal ions and ligand molecules for metalloprotein complexes [58][59].
Unlike proteins, most compounds used in VS are stored in line notation, such as Simplified Molecular Input Line Entry Specification (SMILES) string [60]. The 3D atomic coordinates of these compounds can be obtained from the line notation using several opensource softwares, such as RDKit and Openbabel [61][62][63], or commercial softwares, such as Omega and ConfGen [64][65][66]. Ligand protonation is also important since it affects the net charge of the molecule and the partial charges of individual atoms. Different docking programs will employ different charge assignment protocols. For example, AutoDock uses Gasteiger-Marsili atomic charges whereas the AutoDock Vina does not require the assignment of atomic charges, since the scoring terms that compose its scoring function are charge-independent [67][68].
- (4)
-
After the input files are created, the appropriate docking protocol must be selected. As has been discussed in the section Molecular Docking Protocol, there are many different docking protocols that consider protein and ligand flexibility to enhance the performance of pose prediction. One of the most commonly used protocols is to perform flexible ligand–rigid receptor docking for each docking run, and then dock multiple protein conformations using the ensemble docking strategy [18]. In addition, several docking programs can be combined to avoid the limitations of one algorithm. For instance, Ren and co-workers have explored the effects of using multiple softwares in the pose generation step [69]. They use a RMSD-based criterion to come up with representative poses derived from 3 to 11 different docking programs. The resulting pose prediction achieves better performance than that of each individual docking program.
- (5)
-
Following docking, the results can be rescored or filtered. The computer-generated poses are evaluated based on the ability of the docking protocol to (i) select favorable binding poses for each ligand, and (ii) rank the ligand library to select high scoring hits for experimental measurement. Although the docking calculations are fast enough to process large compound libraries, they suffer from the inherent problem of calculating binding affinities from several simplified scoring terms. One remedy for improving the performance of VS is to employ more rigorous free energy calculations to postprocess docking poses. The main limiting factor in the application of free energy calculations to large chemical libraries is the high computational cost.
In recent years, post-docking filter methods have gained significant interest in drug discovery because they usually provide higher hit rates in VS with low additional computational cost and result in better correlation with experimental data in retrospective benchmarks. Several methods have been designed to eliminate false positive hits obtained from the initial docking experiments. Marcou and co-worker proposed the use of molecular interaction fingerprints (IFP), which are simple bit strings that convert the 3D information of protein–ligand interactions into a 1D vector representation, for the screening of CDK2 inhibitors [70]. The authors demonstrate that using post-docking filters that calculate the Tanimoto similarity of IFP between docked pose and co-crystal pose is more statistically accurate compared to classical scoring functions in discriminating active compounds from inactive ones. They base this on the assumption that active compounds should have certain specific interactions or contacts with their target to display activity. Bertho and co-workers reported a similar post-docking filtering strategy, namely automatically analyzing poses using self-organizing map (AuPoseSOM) to examine the interatomic contacts between the ligand and the target [71]. This type of approach is target-specific and requires the co-crystal ligand pose as the reference. ML can also be applied to this task. Stafford and co-workers introduced AtomNet PoseRanker, a graph CNN trained on PDBbind v2019 to rerank putative co-crystal poses [28].
Another post-docking strategy is the rescoring of docked poses using a consensus model or an advanced ML scoring function. On one hand, the consensus model uses several different scoring functions to re-assess the docking poses generated from a single docking algorithm. Charifson and co-workers have proposed an approach that takes the intersection of the top-scoring molecules according to two or three different scoring functions. They found it provides a dramatic reduction in the number of false positives identified by individual scoring functions on case studies of p38, IMPDH and HIV protease [72]. On the other hand, advanced ML scoring functions developed in recent years, such as AtomNet [73], vScreenML [74], ΔVinaRF20 [75], ΔVinaXGB [76], SIEVE-Score [77] and RF-Score-VS [78], outperform classical scoring functions in screening performance comparisons on benchmark test sets. However, there is no guarantee that ML scoring functions can outperform classical scoring functions on novel targets that are largely different from the samples in the training data set [79].
The above (1) to (5) steps summarize the workflow of VS process. Other structure-based approaches, such as MD simulations, have also been widely utilized in combination with docking to improve VS performance. MD simulations are an efficient approach to discover cryptic binding pockets (in step 2, binding site detection) [49][80], to sample multiple receptor conformations in ensemble docking (in step 4, docking protocols) [15], and to evaluate the interactions of the predicted receptor–ligand complexes (in step 5, post-docking analysis) [81][82].
References
- Maia, E.H.B.; Assis, L.C.; De Oliveira, T.A.; Da Silva, A.M.; Taranto, A.G. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 2020, 8, 343.
- Li, H.; Sze, K.H.; Lu, G.; Ballester, P.J. Machine-learning scoring functions for structure-based virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1478.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444.
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
- Baek, M.; Baker, D. Deep learning and protein structure modeling. Nat. Methods 2022, 19, 13–14.
- Frye, L.; Bhat, S.; Akinsanya, K.; Abel, R. From computer-aided drug discovery to computer-driven drug discovery. Drug Discover. Today Technol. 2021, 39, 111–117.
- Koshland Jr, D.E. The key–lock theory and the induced fit theory. Angew. Chem. Int. Ed. Engl. 1995, 33, 2375–2378.
- Ma, D.-L.; Chan, D.S.-H.; Leung, C.-H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141.
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein–ligand docking. Proteins: Struct. Funct. Bioinform. 1999, 37, 228–241.
- Kearsley, S.K.; Underwood, D.J.; Sheridan, R.P.; Miller, M.D. Flexibases: A way to enhance the use of molecular docking methods. J. Comput. Aided Mol. Des. 1994, 8, 565–582.
- Hart, T.N.; Read, R.J. A multiple-start Monte Carlo docking method. Proteins: Struct. Funct. Bioinform. 1992, 13, 206–222.
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662.
- Wong, C.F. Flexible receptor docking for drug discovery. Expert Opin. Drug Discov. 2015, 10, 1189–1200.
- Tian, S.; Sun, H.; Pan, P.; Li, D.; Zhen, X.; Li, Y.; Hou, T. Assessing an ensemble docking-based virtual screening strategy for kinase targets by considering protein flexibility. J. Chem. Inf. Model. 2014, 54, 2664–2679.
- Korb, O.; Olsson, T.S.; Bowden, S.J.; Hall, R.J.; Verdonk, M.L.; Liebeschuetz, J.W.; Cole, J.C. Potential and limitations of ensemble docking. J. Chem. Inf. Model. 2012, 52, 1262–1274.
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble docking in drug discovery. Biophys. J. 2018, 114, 2271–2278.
- Totrov, M.; Abagyan, R. Flexible ligand docking to multiple receptor conformations: A practical alternative. Curr. Opin. Struct. Biol. 2008, 18, 178–184.
- Rueda, M.; Bottegoni, G.; Abagyan, R. Recipes for the selection of experimental protein conformations for virtual screening. J. Chem. Inf. Model. 2010, 50, 186–193.
- Mohammadi, S.; Narimani, Z.; Ashouri, M.; Firouzi, R.; Karimi-Jafari, M.H. Ensemble learning from ensemble docking: Revisiting the optimum ensemble size problem. Sci. Rep. 2022, 12, 1–15.
- Chandak, T.; Mayginnes, J.P.; Mayes, H.; Wong, C.F. Using machine learning to improve ensemble docking for drug discovery. Proteins: Struct. Funct. Bioinform. 2020, 88, 1263–1270.
- Huang, S.-Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034.
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comp. Biol. 2015, 11, e1004586.
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144.
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. Protein Crystallogr. 2017, 627–641.
- Lee, J.; Freddolino, P.L.; Zhang, Y. Ab initio protein structure prediction. In From Protein Structure to Function with Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 3–35.
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303.
- Stafford, K.A.; Anderson, B.M.; Sorenson, J.; van den Bedem, H. AtomNet PoseRanker: Enriching Ligand Pose Quality for Dynamic Proteins in Virtual High-Throughput Screens. J. Chem. Inf. Model. 2022, 62, 1178–1189.
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229.
- Rollinger, J.M.; Stuppner, H.; Langer, T. Virtual screening for the discovery of bioactive natural products. Nat. Compd. Drugs Vol. I 2008, 211–249.
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337.
- Irwin, J.J.; Shoichet, B.K. ZINC− a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182.
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082.
- Cuesta, S.A.; Mora, J.R.; Márquez, E.A. In silico screening of the DrugBank database to search for possible drugs against SARS-CoV-2. Molecules 2021, 26, 1100.
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672.
- Wishart, D.S.; Tzur, D.; Knox, C.; Eisner, R.; Guo, A.C.; Young, N.; Cheng, D.; Jewell, K.; Arndt, D.; Sawhney, S. HMDB: The human metabolome database. Nucleic Acids Res. 2007, 35, D521–D526.
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617.
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631.
- Sardanelli, A.M.; Isgrò, C.; Palese, L.L. SARS-CoV-2 main protease active site ligands in the human metabolome. Molecules 2021, 26, 1409.
- Liu, Y.; Grimm, M.; Dai, W.-t.; Hou, M.-c.; Xiao, Z.-X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein–ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144.
- Zhang, W.; Bell, E.W.; Yin, M.; Zhang, Y. EDock: Blind protein–ligand docking by replica-exchange monte carlo simulation. J. Cheminform. 2020, 12, 1–17.
- Rooklin, D.; Wang, C.; Katigbak, J.; Arora, P.S.; Zhang, Y. AlphaSpace: Fragment-centric topographical mapping to target protein–protein interaction interfaces. J. Chem. Inf. Model. 2015, 55, 1585–1599.
- Katigbak, J.; Li, H.; Rooklin, D.; Zhang, Y. AlphaSpace 2.0: Representing Concave Biomolecular Surfaces Using β-Clusters. J. Chem. Inf. Model. 2020, 60, 1494–1508.
- Ngan, C.H.; Bohnuud, T.; Mottarella, S.E.; Beglov, D.; Villar, E.A.; Hall, D.R.; Kozakov, D.; Vajda, S. FTMAP: Extended protein mapping with user-selected probe molecules. Nucleic Acids Res. 2012, 40, W271–W275.
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285.
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tuffï¿ ½ry, P. Fpocket: Online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010, 38, W582–W589.
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389.
- Wagner, J.R.; Lee, C.T.; Durrant, J.D.; Malmstrom, R.D.; Feher, V.A.; Amaro, R.E. Emerging computational methods for the rational discovery of allosteric drugs. Chem. Rev. 2016, 116, 6370–6390.
- Oleinikovas, V.; Saladino, G.; Cossins, B.P.; Gervasio, F.L. Understanding cryptic pocket formation in protein targets by enhanced sampling simulations. JACS 2016, 138, 14257–14263.
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein–ligand complexes. Proteins: Struct. Funct. Bioinform. 2008, 73, 765–783.
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating p K prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541.
- Ten Brink, T.; Exner, T.E. pKa based protonation states and microspecies for protein—Ligand docking. J. Comput. Aided Mol. Des. 2010, 24, 935–942.
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667.
- Dolinsky, T.J.; Czodrowski, P.; Li, H.; Nielsen, J.E.; Jensen, J.H.; Klebe, G.; Baker, N.A. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, W522–W525.
- Lie, M.A.; Thomsen, R.; Pedersen, C.N.; Schiøtt, B.; Christensen, M.H. Molecular docking with ligand attached water molecules. J. Chem. Inf. Model. 2011, 51, 909–917.
- Kumar, A.; Zhang, K.Y. Investigation on the effect of key water molecules on docking performance in CSARdock exercise. J. Chem. Inf. Model. 2013, 53, 1880–1892.
- Murphy, R.B.; Repasky, M.P.; Greenwood, J.R.; Tubert-Brohman, I.; Jerome, S.; Annabhimoju, R.; Boyles, N.A.; Schmitz, C.D.; Abel, R.; Farid, R. WScore: A flexible and accurate treatment of explicit water molecules in ligand—Receptor docking. J. Med. Chem. 2016, 59, 4364–4384.
- Yang, C.; Zhang, Y. Lin_F9: A Linear Empirical Scoring Function for Protein–Ligand Docking. J. Chem. Inf. Model. 2021, 61, 4630–4644.
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379.
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36.
- Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. 2013.
- Bento, A.P.; Hersey, A.; Félix, E.; Landrum, G.; Gaulton, A.; Atkinson, F.; Bellis, L.J.; De Veij, M.; Leach, A.R. An open source chemical structure curation pipeline using RDKit. J. Cheminform. 2020, 12, 1–16.
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14.
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584.
- Hawkins, P.C.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936.
- Watts, K.S.; Dalal, P.; Murphy, R.B.; Sherman, W.; Friesner, R.A.; Shelley, J.C. ConfGen: A conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 2010, 50, 534–546.
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461.
- Huey, R.; Morris, G.M. Using AutoDock 4 with AutoDocktools: A tutorial. Scripps Res. Inst. USA 2008, 8, 54–56.
- Ren, X.; Shi, Y.-S.; Zhang, Y.; Liu, B.; Zhang, L.-H.; Peng, Y.-B.; Zeng, R. Novel consensus docking strategy to improve ligand pose prediction. J. Chem. Inf. Model. 2018, 58, 1662–1668.
- Marcou, G.; Rognan, D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207.
- Bouvier, G.; Evrard-Todeschi, N.; Girault, J.-P.; Bertho, G. Automatic clustering of docking poses in virtual screening process using self-organizing map. Bioinformatics 2010, 26, 53–60.
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109.
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv 2015, arXiv:1510.02855.
- Adeshina, Y.O.; Deeds, E.J.; Karanicolas, J. Machine learning classification can reduce false positives in structure-based virtual screening. Proc. Natl. Acad. Sci. USA 2020, 117, 18477–18488.
- Wang, C.; Zhang, Y. Improving scoring-docking-screening powers of protein–ligand scoring functions using random forest. J. Comput. Chem. 2017, 38, 169–177.
- Lu, J.; Hou, X.; Wang, C.; Zhang, Y. Incorporating explicit water molecules and ligand conformation stability in machine-learning scoring functions. J. Chem. Inf. Model. 2019, 59, 4540–4549.
- Yasuo, N.; Sekijima, M. Improved method of structure-based virtual screening via interaction-energy-based learning. J. Chem. Inf. Model. 2019, 59, 1050–1061.
- Wójcikowski, M.; Ballester, P.J.; Siedlecki, P. Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep. 2017, 7, 1–10.
- Su, M.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Tapping on the black box: How is the scoring power of a machine-learning scoring function dependent on the training set? J. Chem. Inf. Model. 2020, 60, 1122–1136.
- Kuzmanic, A.; Bowman, G.R.; Juarez-Jimenez, J.; Michel, J.; Gervasio, F.L. Investigating cryptic binding sites by molecular dynamics simulations. Acc. Chem. Res. 2020, 53, 654–661.
- Sgobba, M.; Caporuscio, F.; Anighoro, A.; Portioli, C.; Rastelli, G. Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 2012, 58, 431–440.
- Kumar, K.; Anbarasu, A.; Ramaiah, S. Molecular docking and molecular dynamics studies on β-lactamases and penicillin binding proteins. Mol. BioSyst. 2014, 10, 891–900.
More
Information
Subjects:
Chemistry, Medicinal
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.4K
Entry Collection:
Biopharmaceuticals Technology
Revisions:
2 times
(View History)
Update Date:
04 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No