Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daria Capece | -- | 2730 | 2022-08-03 11:13:41 | | | |
| 2 | Amina Yu | + 6 word(s) | 2736 | 2022-08-04 04:03:24 | | | | |
| 3 | Amina Yu | -29 word(s) | 2707 | 2022-08-08 02:25:58 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Francesco, B.D.; Verzella, D.; Capece, D.; Vecchiotti, D.; Nolfi, M.D.V.; Flati, I.; Cornice, J.; Padova, M.D.; Angelucci, A.; Alesse, E.; et al. NF-κB in Acute Myeloid Leukemia. Encyclopedia. Available online: https://encyclopedia.pub/entry/25800 (accessed on 25 July 2026).
Francesco BD, Verzella D, Capece D, Vecchiotti D, Nolfi MDV, Flati I, et al. NF-κB in Acute Myeloid Leukemia. Encyclopedia. Available at: https://encyclopedia.pub/entry/25800. Accessed July 25, 2026.
Francesco, Barbara Di, Daniela Verzella, Daria Capece, Davide Vecchiotti, Mauro Di Vito Nolfi, Irene Flati, Jessica Cornice, Monica Di Padova, Adriano Angelucci, Edoardo Alesse, et al. "NF-κB in Acute Myeloid Leukemia" Encyclopedia, https://encyclopedia.pub/entry/25800 (accessed July 25, 2026).
Francesco, B.D., Verzella, D., Capece, D., Vecchiotti, D., Nolfi, M.D.V., Flati, I., Cornice, J., Padova, M.D., Angelucci, A., Alesse, E., & Zazzeroni, F. (2022, August 03). NF-κB in Acute Myeloid Leukemia. In Encyclopedia. https://encyclopedia.pub/entry/25800
Francesco, Barbara Di, et al. "NF-κB in Acute Myeloid Leukemia." Encyclopedia. Web. 03 August, 2022.
Copy Citation
Acute Myeloid Leukemia (AML) is an aggressive hematological malignancy that relies on highly heterogeneous cytogenetic alterations. Although in the last few years new agents have been developed for AML treatment, the overall survival prospects for AML patients are still gloomy and new therapeutic options are still urgently needed. Constitutive NF-κB activation has been reported in around 40% of AML patients, where it sustains AML cell survival and chemoresistance. Given the central role of NF-κB in AML, targeting the NF-κB pathway represents an attractive strategy to treat AML.
NF-κB
acute myeloid leukemia
NF-κB inhibitors
1. NF-κB Pathway in Acute Myeloid Leukemia (AML) Pathogenesis
NF-κB signaling has a central role in AML carcinogenesis, where it is responsible for differentiation, survival, growth of leukemia cells and resistance to therapy [1][2][3]. Indeed, about 40% of patients with AML exhibited increased activity of NF-κB [4]. In particular, it has been demonstrated that NF-κB is constitutively active in CD34+ stem cells from M3, M4 and M5 AML patients [5]. In AML, NF-κB, constitutive activation seems to be crucial for maintaining AML cells (i.e., proliferation and survival) rather than promoting myeloid transformation [2]. In addition, aberrant NF-κB signaling is correlated with resistance of AML cells to radiation and chemotherapy [2]. Evidence has shown that NF-κB mediates chemoresistance in AML, based on its ability to induce anti-apoptotic genes such as BCL-2 (B-cell lymphoma 2) and BCL-XL (B-cell lymphoma-extra-large) [6]. Recently, Wei and collaborators demonstrated that Aurora A-dependent NF-κB signaling drives chemoresistance in AML and was associated with worse clinical outcomes. In particular, it was showed that TRAF-interacting protein with FHA domain (TIFA), which interacts with Tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) to activate the IKK complex, mediated Aurora A-dependent NF-κB activation. In keeping with this observation, TIFA silencing increased chemotoxicities of both AML cells and patient-derived peripheral blood mononuclear cell (PBMC) via inhibition of inflammatory cytokines secretion, leading to tumor necrosis factor α (TNFα)-dependent NF-κB survival pathway attenuation and, thus identifying TIFA as a potential target in AML [7]. Notarbartolo and colleagues demonstrated that NF-κB is important for the establishment of drug resistance in AML by controlling the P-gp (P-glycoprotein)-mediated expression of MDR1 (multidrug resistance mutation 1) gene [8]. Several studies showed that overexpression of heterodimer p50/p65 in resistant variant AML cell lines induced P-gp gene and IAP-family genes expression, underlying the importance of NF-κB proteins in promoting chemoresistance, tumor progression and a poor prognosis [9][10][11].
NF-κB plays a pivotal role also in leukemic stem cells (LSCs), where it regulates survival, proliferation and chemoresistance [12]. Zhou and collaborators demonstrated that NF-κB fosters stem-like properties (i.e., self-renewal capacity) of AML cells via LIN28B activation. Accordingly, NF-κB inhibition reduces LIN28B expression and cell survival as well as LSCs’ self-renewal in vitro, suggesting that inhibition of NF-κB could be a potential opportunity to kill AML cells and LSCs in order to counteract cancer resistance and disease relapse [13].
2. Genetic Alterations Drive NF-κB Constitutive Activation in AML
Chromosomal aberrations involving c-Rel, RelA, NF-κB1 and NF-κB2 genes were found in many hematopoietic and solid tumors. However, in AML, NF-κB constitutive activation ensues from mutations affecting genes involved in the control of NF-κB activity, as detailed below (Figure 1).
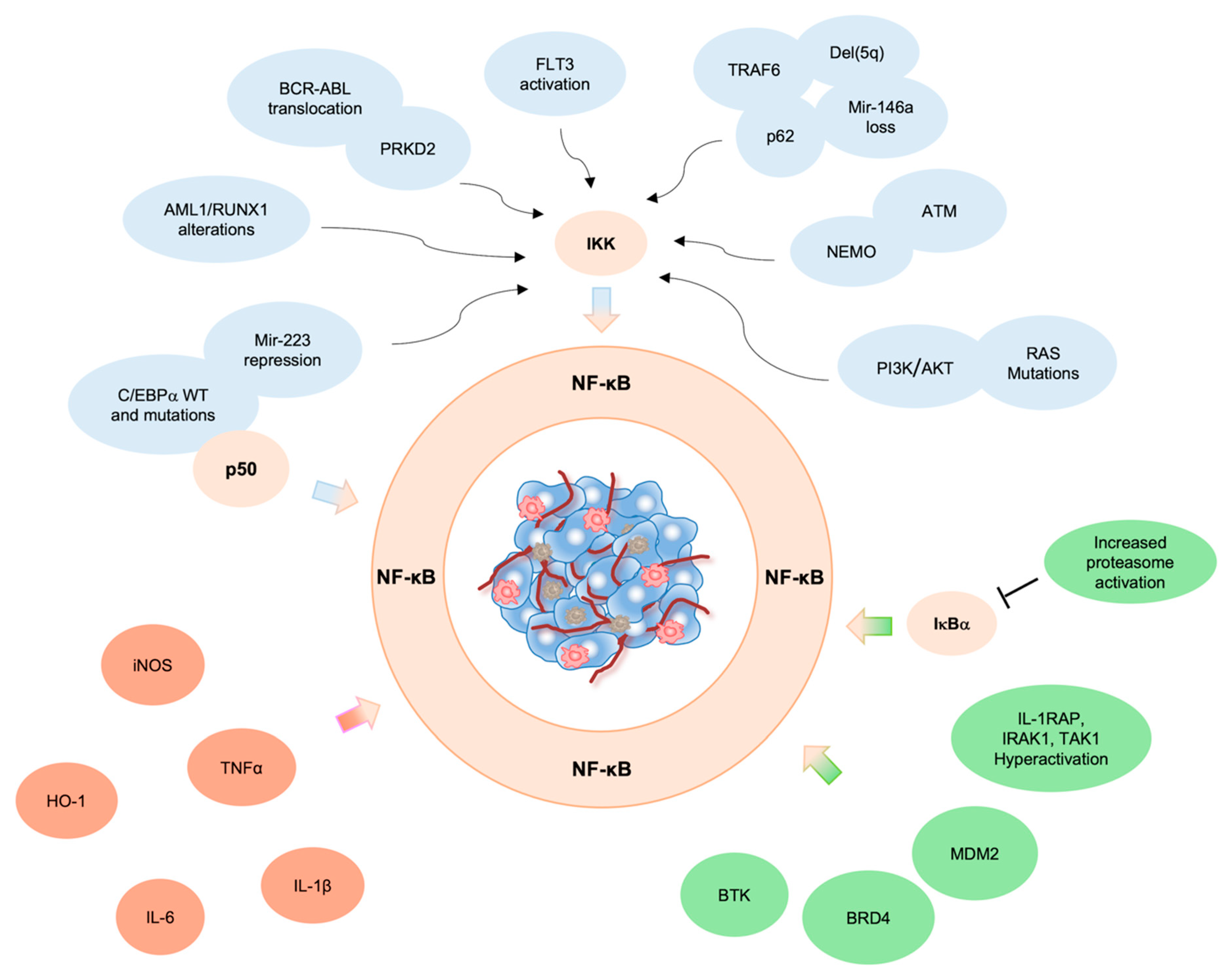
Figure 1. NF-κB activity in AML. Constitutive activation of NF-κB in AML triggered by (i) genetic alterations (blue), (ii) pro-inflammatory cytokines in the TME (fuchsia) and/or (iii) increased expression of NF-κB signaling components (green).
2.1. ATM
Grosjean-Raillard and collaborators demonstrated that activated serine/threonine protein kinase ATM (Ataxia Telangiectasia Mutated), a principal regulator of cell cycle checkpoints in response to DNA damage, triggered IκB kinase NEMO (NF-κB essential modulor) activation and p53-induced death domain protein (PIDD), resulting in NF-κB activation. Pharmacological inhibition of ATM was shown to prevent its autophosphorylation and its interaction with NEMO, leading to redistribution of NEMO and NF-κB to the cytoplasm and apoptosis of malignant myeloblasts [14][15]. These results demonstrate that constitutive phosphorylation of ATM is crucial for NF-κB activation in AML. In addition, it was demonstrated that constitutive activation of Fms related receptor tyrosine kinase 3 (FLT3) signaling resulted in activation of NF-κB via IKK. The inhibition of FLT3 reduced NF-κB activity and promoted apoptosis in AML cell lines and CD34+ primary AML cells [16]. Notably, NEMO-IKK complex promoted the activation of canonical NF-κB pathway in AML [5], thus suggesting that the use of NEMO-binding domain peptides could represent an alternative strategy to indirectly inhibit NF-κB in this cancer.
2.2. BCR/ABL
BCR/ABL translocation, typically found in chronic myeloid leukemia (CML), has been observed also in AML patients. In fact, BCR/ABL+ AML, now classified as a high risk AML, is a rare subtype of AML (0.3–2% of cases) and the prognosis depends on the cytogenetic/molecular landscape [17][18][19]. Studies conducted by different research groups demonstrated that BCR/ABL translocation contributed to IKK-dependent constitutive activation of NF-κB. Accordingly, genetic or pharmacologic inhibition of NF-κB induced cell death in BCR/ABL+ cells [20][21]. it was hypothesized that BCR/ABL fusion protein may activate protein kinase D2 (PRKD2), which in turn induces IKK2-dependent phosphorylation and degradation of IκBα, leading to NF-κB activation [20][21].
2.3. RUNX1
Another transcription factor responsible for aberrant activation of NF-κB in AML is Runt-related transcription factor 1 (RUNX1), also known as AML1. In fact, chromosomal alterations as well as point mutations affecting AML1/RUNX1 gene have been observed in human leukemia, and patients with AML1 mutations exhibited poor clinical outcomes, underlying the important role of RUNX1 during hematopoiesis [22][23][24]. Studies conducted by Nakagawa and collaborators demonstrated that AML1/RUNX1 prevented NF-κB pathway activation in hematopoietic cells by inhibiting IKK activity. Accordingly, AML1 was shown to induce the expression of mir-223, which restrains IKK expression [25][26][27]. In keeping with these observations, mutated AML1/RUNX1 (AML1/ETO fusion product) failed to attenuate IKK kinase activity, leading to aberrant NF-κB activation. Furthermore, pharmacological inhibition of NF-κB signaling reduced tumorigenesis in vivo by suppressing NF-κB-mediated excessive proliferation of AML1/RUNX1 mutated leukemia cells [28].
2.4. C/EBPα
Another important bZIP transcription factor involved in myeloid development and mutated in 10–15% of AML patients is CCAAT/enhancer-binding protein alpha (C/EBPα) [29][30][31]. The presence of double mutations of C/EBPα in AML patients has been associated with a favorable prognosis [32]. Studies conducted by Paz-Priel et al. demonstrated that C/EBPα and its mutated forms interacted with the p50 subunit of NF-κB and induced the espression of NF-κB target genes (i.e., BCL-2 (B-cell lymphoma 2) and c-FLIP (FLICE-like inhibitory protein)) in vitro. In turn, the p50 subunit controlled C/EBPα expression, thus generating a positive feedback loop. it was also showed that these proteins, in complex with p50, can directly regulate the expression of the anti-apoptotic genes BCL-2 and c-FLIP [29][33][34]. Moreover, Pulikkan and collaborators showed that C/EBP transcription factors also regulated IKK expression by inducing the expression of mir-223. Accordingly, repression of mir-223 due to C/EBPα mutation increased IKK-NF-κB activity [35].
2.5. q Deletion
Among genetic mutations, chromosome 5q deletions (del(5q)) are commonly found in high risk myelodysplastic syndromes (MDS)/AML and are associated with favorable prognosis if the percentage of blasts in the bone marrow is less than 5% [36][37]. The main actor implicated in del(5q) is miR-146a, which, under normal conditions, inhibits TRAF6. In vivo studies showed that miR-146a loss in mouse hematopoietic stem and progenitor cells (HSPC) promoted MDS/AML by increasing TRAF6 expression and NF-κB activation [38][39][40]. Pharmacological NF-κB inhibition or genetic inhibition of TRAF6 in AML cells induced G2/M arrest and apoptosis, suggesting that inhibition of TRAF6/NF-κB axis could be a potential approach to treat AML patients with del(5q) and low miR-146a levels. The same results were obtained in miR-146a-depleted HSPC [37]. Further analysis clarified that NF-κB-mediated leukemic cell survival was mediated by sequestosome 1 (SQSTM1, also known as the ubiquitin-binding protein p62), a scaffold protein of the TRAF6/NF-κB axis, whose overexpression is induced by NF-κB, generating a positive feedback loop. Knockdown of p62 resulted in reduced colony formation or tumor growth and reduced TRAF6 activation and NF-κB nuclear localization both in vitro and in vivo, thus indicating that p62 sustains NF-κB activation and leukemic cell functions (i.e., cell cycle and myeloid cell development) through TRAF6. Overall, these findings support the hypothesis that theh TRAF6/p62/NF-κB axis is responsible for leukemic cell survival in del(5q) AML with low miR-146a [37].
2.6. RAS
It is well demonstrated that NF-κB is also activated by N-RAS and K-RAS mutations that occur in approximately 20% of AML cases [41][42][43]. Birkenkamp and collaborators showed an important association between constitutive NF-κB activity and persistent Ras/phosphoinositide 3-kinase (PI3K)/protein kinase B (PKB) signaling in human AML blasts, where these two different signaling pathways sustain the survival of AML cells. Althought activating mutations in FLT3 and c-Kit (receptor tyrosine kinase (RTK)), which encode receptor tyrosine kinases upstream of Ras, were shown to be responsible for aberrant activation of Ras signaling in 40% of AML cases [43], Birken and collaborators demonstrated that NF-κB activation in AML blasts was not dependent on FLT3 mutations, but was triggered by RAS and PI3K/AKT (protein kinase B (PKA)) pathways, as the pharmacological inhibition of these two signaling blocked NF-κB activity [44]. Nevertheless, the role of FLT3 remains controversial, as some reports in the literature showed that FLT3 overexpression, and/or mutations (i.e., FLT3-ITD (internal tandem duplication), the most common genetic abnormality), can induce canonical or non-canonical NF-κB signaling in AML. Accordingly, genetic or pharmacological inhibition of FLT3 reduces NF-κB activity and promotes apoptosis of both AML cell lines and primary cells [16][45][46] (Figure 1).
3. Pro-Inflammatory Microenvironment and NF-κB Activation in Leukemic Cells
Beyond genetic alteration, aberrant NF-κB activation could also stem from the steady exposure of tumor cells to inflammatory stimuli and other cues emanating from the TME.
Inflammation-induced tumorigenesis is one of the most important mechanisms underpinning the proliferation of leukemic cells. It is known that NF-κB is the major player between inflammation and cancer [47][48] and contributes to evading apoptosis and sustaining cell survival by inducing the secretion of pro-inflammatory cytokines (i.e., TNFα, Interleukin 6 (IL-6), Interleukin 1β (IL-1β) and regulating heme oxygenase-1 (HO-1) expression and inducible nitric oxide synthase (iNOS) activation in the TME [49][50][51]. Although the increased activation of iNOS is associated with pro-apoptotic functions, the presence of high iNOS expression in AML patients suggests that it could either promote or inhibit apoptosis during carcinogenesis, probably depending on inflammation status [52][53]. As reported in the literature, a subset of AMLs is characterized by secretion of high levels of pro-inflammatory cytokines, that in turn activate NF-κB pathway, thus generating a positive feedback loop able to sustain NF-κB-dependent AML growth both in patients and in murine models [2]. The role of TME in fostering tumor progression by paracrine secretion of cytokines is well established [54][55]. Jacamo and collaborators demonstrated that bone marrow stromal cells promoted NF-κB activation in AML cells through vascular cell adhesion molecule 1 (VCAM-1) and very late antigen 4 (VLA-4) interaction [56] and highlighted the importance of surrounding stroma in induction and maintenance of aberrant NF-κB activation in tumor cells [57][58][59]. Recent findings showed that the immune modulator Interferon regulatory factor 2 binding protein 2 (IRF2BP2) attenuates the inflammatory signals between monocytic AML cells by controlling the NF-κB-mediated TNFα signaling. Accordingly, the suppression of IRF2BP2 induces caspase 8- and caspase 3-mediated apoptotic cell death via NF-κB mediated upregulation of IL-β1 [60][61]. In keeping with these findings, Volk and collaborators demonstrated that TNF stimulated leukemia cell growth via autocrine induction of NF-κB and JNK-AP1 (c-Jun N-terminal kinases/activator protein-1) signaling, suggesting that several signaling pathways cooperate to sustain cancer cell proliferation. In fact, solely NF-κB inhibition was not sufficient in inhibiting AML tumor growth, due to the activation of anti-apoptotic genes by the TNF/JNK axis. Accordingly, in vivo studies demonstrated that the inhibition of NF-κB along with TNF inactivation induced leukemic cell death. Additionally, it was showed that TNF produced by leukemic cells inhibited normal HSPC growth in a paracrine manner, thus finding a possible explanation for the hematopoietic repression observed in AML patients. These findings suggest that co-inhibition of both TNF/JNK-AP1 and TNF/NF-κB pathways could be a potential therapeutic approach to inhibit leukemic cell growth while protecting HSPC in those AML Fab subtypes which express TNF, such as M3, M4, M5 [62]. An additional study underscoring the paramount role of TNF/NF-κB axis in AML was published by Kagoya and collaborators. It was demonstrated that LSCs, also known as leukemia-initiating cells (LICs), showed constitutive NF-κB activity due to autocrine TNFα secretion, that in turn promoted IκBα degradation through prolonged activation of proteasome machinery, induction of IκBα degradation and translocation of NF-κB into the nucleus. Since TNFα is one of the NF-κB target genes, leukemia progression is maintained by the NF-κB/TNFα feedback loop [63][64]. Conversely, NF-κB inhibition in LICs curbs tumorigenesis in vivo, suggesting that the NF-κB/TNFα axis supports leukemia progression [64]. Furthermore, Li and collaborators demonstrated that treatment with TNF and IL-1β inhibitors sensitized LSCs to NF-κB inhibition indicating that this combination strategy could be used to remove leukemic cells as well as LSCs and overcome the LSCs-mediated drug resistance [65] (Figure 1).
4. Aberrant NF-κB Activation by NF-κB Regulators/Interactors
The hyperactivation of NF-κB in leukemic cells is also due to an increased activation of upstream regulators of the NF-κB pathway [66][67], making these components promising targets for cancer treatment. Several studies identified Interleukin-1 receptor accessory protein (IL-1RAP), Interleukin-1 receptor-associated kinase 1 (IRAK1), transforming growth factor-β activated kinase (TAK1) and Bruton’s tyrosine kinase (BTK) as overexpressed in primary AML cells. Accordingly, inhibition of these proteins suppressed NF-κB activation and restrained tumor growth by promoting apoptosis both in vitro and in vivo [2][68][69][70][71].
The proviral insertion in murine (PIM) lymphoma proteins are proto-oncogenic serine/threonine kinases, constitutively active in AML [72][73]. Recent studies demonstrated that PIM2 supports AML tumorigenesis by suppressing apoptosis and inducing cancer cell survival via NF-κB activation [74]. Indeed, PIM2 activates the NF-κB pathway by inducing the phosphorylation of serine threonine kinase Cot and consequently the upregulation and degradation of IκB. Furthermore, Nihira and collaborators demonstrated that Pim1 controls the NF-κB pathway by phosphorylating RelA/p65 at Ser276 [75]. On the other hand, it has been demonstrated that NF-κB regulates the expression of Pim-1 and 2 kinases through CD40 signaling and in response to FLT3/ITB oncogenic mutants, respectively [74][76].
Other important players promoting constitutive activation of the NF-κB pathway are proteins involved in NF-κB-mediated gene transcription such as Bromodomain-containing protein 4 (BRD4) and murine double minute 2 (MDM2). BRD4 belongs to bromodomain and extra terminal domain (BET) protein family and interacts with acetylated lysine in histone or non-histone proteins [77][78]. BRD4 plays a central role in transcriptional regulation via interactions with specific proteins like positive transcription elongation factor B (pTEFb). With regard to NF-κB pathway, although the molecular mechanism is still poorly understood, BRD4 might bind to acetylated histones and proteins, such as p65, leading to transcription of the NF-κB-regulated pro-inflammatory genes [78][79][80][81]. Silencing of BRD4 reduced leukemic tumor growth in mice [82]. Moreover, pharmacological inhibition of BRD4 with BEF inhibitors restrained NF-κB-mediated inflammatory response [83]. Accordingly, treatment with I-BET762, a pan-BET inhibitor, suppressed inflammation and reduced the incidence of death in mice after prolonged lipopolysaccharide (LPS) exposure [84].
It has been demonstrated that MDM2 induces p65 expression in different cells; MDM2 overexpression was observed in AML, but its capacity to interact with NF-κB remains debated [85][86]. On the other hand, overexpression of negative regulator MDM2 inactivates p53, which is frequently inactivated in AML cell lines and patients and whose loss is associated with poor prognosis. Recently, it has been demonstrated that inhibition of the p53-MDM2 complex promoted antitumor activity in vivo [87].
Constitutive NF-κB expression is also associated with the enhanced activity of an immunoproteasome variant expressed in hematopoietic cells, functionally close to “classical” proteasome, which represents an attractive target in hematologic malignancies [88]. Accordingly, increased activation of this proteasome variant has been observed in CD34+ leukemic cells and in LICs from different murine models [64][89][90][91] (Figure 1).
References
- Labbozzetta, M.; Notarbartolo, M.; Poma, P. Can NF-ΚB Be Considered a Valid Drug Target in Neoplastic Diseases? Our Point of View. Int. J. Mol. Sci. 2020, 21, 3070.
- Bosman, M.C.J.; Schuringa, J.J.; Vellenga, E. Constitutive NF-ΚB Activation in AML: Causes and Treatment Strategies. Crit. Rev. Oncol./Hematol. 2016, 98, 35–44.
- Gasparini, C.; Celeghini, C.; Monasta, L.; Zauli, G. NF-ΚB Pathways in Hematological Malignancies. Cell. Mol. Life Sci. CMLS 2014, 71, 2083–2102.
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear Factor-KappaB Is Constitutively Activated in Primitive Human Acute Myelogenous Leukemia Cells. Blood 2001, 98, 2301–2307.
- Baumgartner, B.; Weber, M.; Quirling, M.; Fischer, C.; Page, S.; Adam, M.; Von Schilling, C.; Waterhouse, C.; Schmid, C.; Neumeier, D.; et al. Increased IkappaB Kinase Activity Is Associated with Activated NF-KappaB in Acute Myeloid Blasts. Leukemia 2002, 16, 2062–2071.
- Mehta, S.V.; Shukla, S.N.; Vora, H.H. Overexpression of Bcl2 Protein Predicts Chemoresistance in Acute Myeloid Leukemia: Its Correlation with FLT3. Neoplasma 2013, 60, 666–675.
- Wei, T.-Y.W.; Wu, P.-Y.; Wu, T.-J.; Hou, H.-A.; Chou, W.-C.; Teng, C.-L.J.; Lin, C.-R.; Chen, J.-M.M.; Lin, T.-Y.; Su, H.-C.; et al. Aurora a and NF-ΚB Survival Pathway Drive Chemoresistance in Acute Myeloid Leukemia via the TRAF-Interacting Protein TIFA. Cancer Res. 2017, 77, 494–508.
- Notarbartolo, M.; Cervello, M.; Dusonchet, L.; Cusimano, A.; D’Alessandro, N. Resistance to Diverse Apoptotic Triggers in Multidrug Resistant HL60 Cells and Its Possible Relationship to the Expression of P-Glycoprotein, Fas and of the Novel Anti-Apoptosis Factors IAP (Inhibitory of Apoptosis Proteins). Cancer Lett. 2002, 180, 91–101.
- Notarbartolo, M.; Cervello, M.; Giannitrapani, L.; Meli, M.; Poma, P.; Dusonchet, L.; Montalto, G.; D’Alessandro, N. Expression of IAPs and Alternative Splice Variants in Hepatocellular Carcinoma Tissues and Cells. Ann. N. Y. Acad. Sci. 2004, 1028, 289–293.
- Tamm, I.; Kornblau, S.M.; Segall, H.; Krajewski, S.; Welsh, K.; Kitada, S.; Scudiero, D.A.; Tudor, G.; Qui, Y.H.; Monks, A.; et al. Expression and Prognostic Significance of IAP-Family Genes in Human Cancers and Myeloid Leukemias. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 1796–1803.
- Hrdinka, M.; Yabal, M. Inhibitor of Apoptosis Proteins in Human Health and Disease. Genes Immun. 2019, 20, 641–650.
- Marchand, T.; Pinho, S. Leukemic Stem Cells: From Leukemic Niche Biology to Treatment Opportunities. Front. Immunol. 2021, 12, 775128.
- Zhou, J.; Chooi, J.Y.; Ching, Y.Q.; Quah, J.Y.; Toh, S.H.M.; Ng, Y.; Tan, T.Z.; Chng, W.J. NF-ΚB Promotes the Stem-like Properties of Leukemia Cells by Activation of LIN28B. World J. Stem Cells 2018, 10, 34–42.
- Grosjean-Raillard, J.; Tailler, M.; Adès, L.; Perfettini, J.-L.; Fabre, C.; Braun, T.; De Botton, S.; Fenaux, P.; Kroemer, G. ATM Mediates Constitutive NF-KappaB Activation in High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia. Oncogene 2009, 28, 1099–1109.
- Shiloh, Y.; Ziv, Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210.
- Grosjean-Raillard, J.; Adès, L.; Boehrer, S.; Tailler, M.; Fabre, C.; Braun, T.; De Botton, S.; Israel, A.; Fenaux, P.; Kroemer, G. Flt3 Receptor Inhibition Reduces Constitutive NFkappaB Activation in High-Risk Myelodysplastic Syndrome and Acute Myeloid Leukemia. Apoptosis Int. J. Program. Cell Death 2008, 13, 1148–1161.
- Neuendorff, N.R.; Burmeister, T.; Dörken, B.; Westermann, J. BCR-ABL-Positive Acute Myeloid Leukemia: A New Entity? Analysis of Clinical and Molecular Features. Ann. Hematol. 2016, 95, 1211–1221.
- Neuendorff, N.R.; Hemmati, P.; Arnold, R.; Ihlow, J.; Dörken, B.; Müller-Tidow, C.; Westermann, J. BCR-ABL(+) Acute Myeloid Leukemia: Are We Always Dealing with a High-Risk Disease? Blood Adv. 2018, 2, 1409–1411.
- Mariotti, B.; Meconi, F.; Palmieri, R.; De Bellis, E.; Lavorgna, S.; Ottone, T.; Martini, V.; Lo-Coco, F.; Cicconi, L. Acute Myeloid Leukemia with Concomitant BCR-ABL and NPM1 Mutations. Case Rep. Hematol. 2019, 2019, 6707506.
- Hsieh, M.-Y.; Van Etten, R.A. IKK-Dependent Activation of NF-ΚB Contributes to Myeloid and Lymphoid Leukemogenesis by BCR-ABL1. Blood 2014, 123, 2401–2411.
- Mihailovic, T.; Marx, M.; Auer, A.; Van Lint, J.; Schmid, M.; Weber, C.; Seufferlein, T. Protein Kinase D2 Mediates Activation of Nuclear Factor KappaB by Bcr-Abl in Bcr-Abl+ Human Myeloid Leukemia Cells. Cancer Res. 2004, 64, 8939–8944.
- Schnittger, S.; Dicker, F.; Kern, W.; Wendland, N.; Sundermann, J.; Alpermann, T.; Haferlach, C.; Haferlach, T. RUNX1 Mutations Are Frequent in de Novo AML with Noncomplex Karyotype and Confer an Unfavorable Prognosis. Blood 2011, 117, 2348–2357.
- Miyoshi, H.; Shimizu, K.; Kozu, T.; Maseki, N.; Kaneko, Y.; Ohki, M. T(8;21) Breakpoints on Chromosome 21 in Acute Myeloid Leukemia Are Clustered within a Limited Region of a Single Gene, AML1. Proc. Natl. Acad. Sci. USA 1991, 88, 10431–10434.
- Marcucci, G.; Caligiuri, M.A.; Bloomfield, C.D. Molecular and Clinical Advances in Core Binding Factor Primary Acute Myeloid Leukemia: A Paradigm for Translational Research in Malignant Hematology. Cancer Investig. 2000, 18, 768–780.
- Fazi, F.; Racanicchi, S.; Zardo, G.; Starnes, L.M.; Mancini, M.; Travaglini, L.; Diverio, D.; Ammatuna, E.; Cimino, G.; Lo-Coco, F.; et al. Epigenetic Silencing of the Myelopoiesis Regulator MicroRNA-223 by the AML1/ETO Oncoprotein. Cancer Cell 2007, 12, 457–466.
- Fukao, T.; Fukuda, Y.; Kiga, K.; Sharif, J.; Hino, K.; Enomoto, Y.; Kawamura, A.; Nakamura, K.; Takeuchi, T.; Tanabe, M. An Evolutionarily Conserved Mechanism for MicroRNA-223 Expression Revealed by MicroRNA Gene Profiling. Cell 2007, 129, 617–631.
- Li, T.; Morgan, M.J.; Choksi, S.; Zhang, Y.; Kim, Y.-S.; Liu, Z. MicroRNAs Modulate the Noncanonical Transcription Factor NF-KappaB Pathway by Regulating Expression of the Kinase IKKalpha during Macrophage Differentiation. Nat. Immunol. 2010, 11, 799–805.
- Nakagawa, M.; Shimabe, M.; Watanabe-Okochi, N.; Arai, S.; Yoshimi, A.; Shinohara, A.; Nishimoto, N.; Kataoka, K.; Sato, T.; Kumano, K.; et al. AML1/RUNX1 Functions as a Cytoplasmic Attenuator of NF-ΚB Signaling in the Repression of Myeloid Tumors. Blood 2011, 118, 6626–6637.
- Paz-Priel, I.; Friedman, A. C/EBPα Dysregulation in AML and ALL. Crit. Rev. Oncog. 2011, 16, 93–102.
- Roe, J.-S.; Vakoc, C.R. C/EBPα: Critical at the Origin of Leukemic Transformation. J. Exp. Med. 2014, 211, 1–4.
- Grardel, N.; Roumier, C.; Soenen, V.; Lai, J.L.; Plantier, I.; Gheveart, C.; Cosson, A.; Fenaux, P.; Preudhomme, C. Acute Myeloblastic Leukemia (AML) with Inv (16)(P13;Q22) and the Rare I Type CBFbeta-MYH11 Transcript: Report of Two New Cases. Leukemia 2002, 16, 150–151.
- Green, C.L.; Koo, K.K.; Hills, R.K.; Burnett, A.K.; Linch, D.C.; Gale, R.E. Prognostic Significance of CEBPA Mutations in a Large Cohort of Younger Adult Patients with Acute Myeloid Leukemia: Impact of Double CEBPA Mutations and the Interaction with FLT3 and NPM1 Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 2739–2747.
- Paz-Priel, I.; Cai, D.H.; Wang, D.; Kowalski, J.; Blackford, A.; Liu, H.; Heckman, C.A.; Gombart, A.F.; Koeffler, H.P.; Boxer, L.M.; et al. CCAAT/Enhancer Binding Protein Alpha (C/EBPalpha) and C/EBPalpha Myeloid Oncoproteins Induce Bcl-2 via Interaction of Their Basic Regions with Nuclear Factor-KappaB P50. Mol. Cancer Res. MCR 2005, 3, 585–596.
- Paz-Priel, I.; Ghosal, A.K.; Kowalski, J.; Friedman, A.D. C/EBPalpha or C/EBPalpha Oncoproteins Regulate the Intrinsic and Extrinsic Apoptotic Pathways by Direct Interaction with NF-KappaB P50 Bound to the Bcl-2 and FLIP Gene Promoters. Leukemia 2009, 23, 365–374.
- Pulikkan, J.A.; Peramangalam, P.S.; Dengler, V.; Ho, P.A.; Preudhomme, C.; Meshinchi, S.; Christopeit, M.; Nibourel, O.; Müller-Tidow, C.; Bohlander, S.K.; et al. C/EBPα Regulated MicroRNA-34a Targets E2F3 during Granulopoiesis and Is down-Regulated in AML with CEBPA Mutations. Blood 2010, 116, 5638–5649.
- Ebert, B.L. Deletion 5q in Myelodysplastic Syndrome: A Paradigm for the Study of Hemizygous Deletions in Cancer. Leukemia 2009, 23, 1252–1256.
- Fang, J.; Barker, B.; Bolanos, L.; Liu, X.; Jerez, A.; Makishima, H.; Christie, S.; Chen, X.; Rao, D.S.; Grimes, H.L.; et al. Myeloid Malignancies with Chromosome 5q Deletions Acquire a Dependency on an Intrachromosomal NF-ΚB Gene Network. Cell Rep. 2014, 8, 1328–1338.
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-KappaB-Dependent Induction of MicroRNA MiR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486.
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. MiR-146a Is a Significant Brake on Autoimmunity, Myeloproliferation, and Cancer in Mice. J. Exp. Med. 2011, 208, 1189–1201.
- Zhao, J.L.; Rao, D.S.; Boldin, M.P.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. NF-KappaB Dysregulation in MicroRNA-146a-Deficient Mice Drives the Development of Myeloid Malignancies. Proc. Natl. Acad. Sci. USA 2011, 108, 9184–9189.
- Beaupre, D.M.; Kurzrock, R. RAS and Leukemia: From Basic Mechanisms to Gene-Directed Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1999, 17, 1071–1079.
- Reuter, C.W.; Morgan, M.A.; Bergmann, L. Targeting the Ras Signaling Pathway: A Rational, Mechanism-Based Treatment for Hematologic Malignancies? Blood 2000, 96, 1655–1669.
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting Oncogenic Ras Signaling in Hematologic Malignancies. Blood 2012, 120, 3397–3406.
- Birkenkamp, K.U.; Geugien, M.; Schepers, H.; Westra, J.; Lemmink, H.H.; Vellenga, E. Constitutive NF-KappaB DNA-Binding Activity in AML Is Frequently Mediated by a Ras/PI3-K/PKB-Dependent Pathway. Leukemia 2004, 18, 103–112.
- Takahashi, S.; Harigae, H.; Ishii, K.K.; Inomata, M.; Fujiwara, T.; Yokoyama, H.; Ishizawa, K.; Kameoka, J.; Licht, J.D.; Sasaki, T.; et al. Over-Expression of Flt3 Induces NF-KappaB Pathway and Increases the Expression of IL-6. Leuk. Res. 2005, 29, 893–899.
- Shanmugam, R.; Gade, P.; Wilson-Weekes, A.; Sayar, H.; Suvannasankha, A.; Goswami, C.; Li, L.; Gupta, S.; Cardoso, A.A.; Baghdadi, T.A.; et al. A Noncanonical Flt3ITD/NF-ΚB Signaling Pathway Represses DAPK1 in Acute Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 360–369.
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41.
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899.
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and Colon Cancer. Gastroenterology 2010, 138, 2101–2114.e5.
- Rushworth, S.A.; MacEwan, D.J. HO-1 Underlies Resistance of AML Cells to TNF-Induced Apoptosis. Blood 2008, 111, 3793–3801.
- Heasman, S.-A.; Zaitseva, L.; Bowles, K.M.; Rushworth, S.A.; Macewan, D.J. Protection of Acute Myeloid Leukaemia Cells from Apoptosis Induced by Front-Line Chemotherapeutics Is Mediated by Haem Oxygenase-1. Oncotarget 2011, 2, 658–668.
- Olson, S.Y.; Garbán, H.J. Regulation of Apoptosis-Related Genes by Nitric Oxide in Cancer. Nitric Oxide Biol. Chem. 2008, 19, 170–176.
- Brandão, M.M.; Soares, E.; Salles, T.S.; Saad, S.T. Expression of Inducible Nitric Oxide Synthase Is Increased in Acute Myeloid Leukaemia. Acta Haematol. 2001, 106, 95–99.
- Capece, D.; D’Andrea, D.; Verzella, D.; Tornatore, L.; Begalli, F.; Bennett, J.; Zazzeroni, F.; Franzoso, G. Turning an Old GADDget into a Troublemaker. Cell Death Differ. 2018, 25, 642–644.
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.-U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered Microenvironmental Regulation of Leukemic and Normal Stem Cells in Chronic Myelogenous Leukemia. Cancer Cell 2012, 21, 577–592.
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal Leukemia-Stroma VCAM-1/VLA-4-Dependent Activation of NF-ΚB Mediates Chemoresistance. Blood 2014, 123, 2691–2702.
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209.
- Park, M.H.; Hong, J.T. Roles of NF-ΚB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15.
- Vlahopoulos, S.A. Aberrant Control of NF-ΚB in Cancer Permits Transcriptional and Phenotypic Plasticity, to Curtail Dependence on Host Tissue: Molecular Mode. Cancer Biol. Med. 2017, 14, 254–270.
- Puissant, A.; Medyouf, H. Walking the Tightrope: Balancing Delicate Inflammation Response to Eradicate Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 1617–1619.
- Ellegast, J.M.; Alexe, G.; Hamze, A.; Lin, S.; Uckelmann, H.J.; Rauch, P.J.; Pimkin, M.; Ross, L.S.; Dharia, N.V.; Robichaud, A.L.; et al. Unleashing Cell-Intrinsic Inflammation as a Strategy to Kill AML Blasts. Cancer Discov. 2022, 12, 1760–1781.
- Volk, A.; Li, J.; Xin, J.; You, D.; Zhang, J.; Liu, X.; Xiao, Y.; Breslin, P.; Li, Z.; Wei, W.; et al. Co-Inhibition of NF-ΚB and JNK Is Synergistic in TNF-Expressing Human AML. J. Exp. Med. 2014, 211, 1093–1108.
- Csizmar, C.M.; Kim, D.-H.; Sachs, Z. The Role of the Proteasome in AML. Blood Cancer J. 2016, 6, e503.
- Kagoya, Y.; Yoshimi, A.; Kataoka, K.; Nakagawa, M.; Kumano, K.; Arai, S.; Kobayashi, H.; Saito, T.; Iwakura, Y.; Kurokawa, M. Positive Feedback between NF-ΚB and TNF-α Promotes Leukemia-Initiating Cell Capacity. J. Clin. Investig. 2014, 124, 528–542.
- Li, J.; Volk, A.; Zhang, J.; Cannova, J.; Dai, S.; Hao, C.; Hu, C.; Sun, J.; Xu, Y.; Wei, W.; et al. Sensitizing Leukemia Stem Cells to NF-ΚB Inhibitor Treatment in Vivo by Inactivation of Both TNF and IL-1 Signaling. Oncotarget 2016, 8, 8420–8435.
- Grondona, P.; Bucher, P.; Schulze-Osthoff, K.; Hailfinger, S.; Schmitt, A. NF-ΚB Activation in Lymphoid Malignancies: Genetics, Signaling, and Targeted Therapy. Biomedicines 2018, 6, 38.
- Xiao, G.; Fu, J. NF-ΚB and Cancer: A Paradigm of Yin-Yang. Am. J. Cancer Res. 2011, 1, 192–221.
- Fang, J.; Rhyasen, G.; Bolanos, L.; Rasch, C.; Varney, M.; Wunderlich, M.; Goyama, S.; Jansen, G.; Cloos, J.; Rigolino, C.; et al. Cytotoxic Effects of Bortezomib in Myelodysplastic Syndrome/Acute Myeloid Leukemia Depend on Autophagy-Mediated Lysosomal Degradation of TRAF6 and Repression of PSMA1. Blood 2012, 120, 858–867.
- Hosseini, M.M.; Kurtz, S.E.; Abdelhamed, S.; Mahmood, S.; Davare, M.A.; Kaempf, A.; Elferich, J.; McDermott, J.E.; Liu, T.; Payne, S.H.; et al. Inhibition of Interleukin-1 Receptor-Associated Kinase-1 Is a Therapeutic Strategy for Acute Myeloid Leukemia Subtypes. Leukemia 2018, 32, 2374–2387.
- Bosman, M.C.J.; Schepers, H.; Jaques, J.; Brouwers-Vos, A.Z.; Quax, W.J.; Schuringa, J.J.; Vellenga, E. The TAK1-NF-ΚB Axis as Therapeutic Target for AML. Blood 2014, 124, 3130–3140.
- Rhyasen, G.W.; Bolanos, L.; Starczynowski, D.T. Differential IRAK Signaling in Hematologic Malignancies. Exp. Hematol. 2013, 41, 1005–1007.
- Sawaguchi, Y.; Yamazaki, R.; Nishiyama, Y.; Mae, M.; Abe, A.; Nishiyama, H.; Nishisaka, F.; Ibuki, T.; Sasai, T.; Matsuzaki, T. Novel Pan-Pim Kinase Inhibitors with Imidazopyridazine and Thiazolidinedione Structure Exert Potent Antitumor Activities. Front. Pharmacol. 2021, 12, 672536.
- Liu, Z.; Han, M.; Ding, K.; Fu, R. The Role of Pim Kinase in Immunomodulation. Am. J. Cancer Res. 2020, 10, 4085–4097.
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein Kinase PIM2: A Simple PIM Family Kinase with Complex Functions in Cancer Metabolism and Therapeutics. J. Cancer 2021, 12, 2570–2581.
- Nihira, K.; Ando, Y.; Yamaguchi, T.; Kagami, Y.; Miki, Y.; Yoshida, K. Pim-1 Controls NF-B Signalling by Stabilizing RelA/P65. Cell Death Differ. 2010, 17, 689–698.
- Zhu, N.; Ramirez, L.M.; Lee, R.L.; Magnuson, N.S.; Bishop, G.A.; Gold, M.R. CD40 Signaling in B Cells Regulates the Expression of the Pim-1 Kinase via the NF-ΚB Pathway. J. Immunol. 2002, 168, 744–754.
- Filippakopoulos, P.; Knapp, S. Targeting Bromodomains: Epigenetic Readers of Lysine Acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356.
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFκB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16.
- Huang, B.; Yang, X.-D.; Zhou, M.-M.; Ozato, K.; Chen, L.-F. Brd4 Coactivates Transcriptional Activation of NF-KappaB via Specific Binding to Acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387.
- Zou, Z.; Huang, B.; Wu, X.; Zhang, H.; Qi, J.; Bradner, J.; Nair, S.; Chen, L.-F. Brd4 Maintains Constitutively Active NF-ΚB in Cancer Cells by Binding to Acetylated RelA. Oncogene 2014, 33, 2395–2404.
- Smale, S.T. Hierarchies of NF-ΚB Target-Gene Regulation. Nat. Immunol. 2011, 12, 689–694.
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi Screen Identifies Brd4 as a Therapeutic Target in Acute Myeloid Leukaemia. Nature 2011, 478, 524–528.
- Brown, J.D.; Lin, C.Y.; Duan, Q.; Griffin, G.; Federation, A.; Paranal, R.M.; Bair, S.; Newton, G.; Lichtman, A.; Kung, A.; et al. NF-ΚB Directs Dynamic Super Enhancer Formation in Inflammation and Atherogenesis. Mol. Cell 2014, 56, 219–231.
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of Inflammation by a Synthetic Histone Mimic. Nature 2010, 468, 1119–1123.
- Gu, L.; Findley, H.W.; Zhou, M. MDM2 Induces NF-KappaB/P65 Expression Transcriptionally through Sp1-Binding Sites: A Novel, P53-Independent Role of MDM2 in Doxorubicin Resistance in Acute Lymphoblastic Leukemia. Blood 2002, 99, 3367–3375.
- Thomasova, D.; Mulay, S.R.; Bruns, H.; Anders, H.-J. P53-Independent Roles of MDM2 in NF-ΚB Signaling: Implications for Cancer Therapy, Wound Healing, and Autoimmune Diseases. Neoplasia 2012, 14, 1097–1101.
- Hayashi, Y.; Goyama, S.; Liu, X.; Tamura, M.; Asada, S.; Tanaka, Y.; Fukuyama, T.; Wunderlich, M.; O’Brien, E.; Mizukawa, B.; et al. Antitumor Immunity Augments the Therapeutic Effects of P53 Activation on Acute Myeloid Leukemia. Nat. Commun. 2019, 10, 4869.
- Kuhn, D.J.; Orlowski, R.Z. The Immunoproteasome as a Target in Hematologic Malignancies. Semin. Hematol. 2012, 49, 258–262.
- Ma, W.; Kantarjian, H.; Bekele, B.; Donahue, A.C.; Zhang, X.; Zhang, Z.J.; O’Brien, S.; Estey, E.; Estrov, Z.; Cortes, J.; et al. Proteasome Enzymatic Activities in Plasma as Risk Stratification of Patients with Acute Myeloid Leukemia and Advanced-Stage Myelodysplastic Syndrome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3820–3826.
- Niewerth, D.; Dingjan, I.; Cloos, J.; Jansen, G.; Kaspers, G. Proteasome Inhibitors in Acute Leukemia. Expert Rev. Anticancer Ther. 2013, 13, 327–337.
- Bonardi, F.; Fusetti, F.; Deelen, P.; van Gosliga, D.; Vellenga, E.; Schuringa, J.J. A Proteomics and Transcriptomics Approach to Identify Leukemic Stem Cell (LSC) Markers. Mol. Cell. Proteom. MCP 2013, 12, 626–637.
More
Information
Subjects:
Pathology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
924
Revisions:
3 times
(View History)
Update Date:
08 Aug 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No