Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Salman Ahmed | -- | 5910 | 2022-07-28 15:28:15 | | | |
| 2 | Catherine Yang | -3822 word(s) | 2088 | 2022-07-29 03:09:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ahmed, S.; Alam, W.; Jeandet, P.; Aschner, M.; Alsharif, K.F.; Saso, L.; Khan, H. Marine Peptides in Prostate Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/25616 (accessed on 22 July 2026).
Ahmed S, Alam W, Jeandet P, Aschner M, Alsharif KF, Saso L, et al. Marine Peptides in Prostate Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/25616. Accessed July 22, 2026.
Ahmed, Salman, Waqas Alam, Philippe Jeandet, Michael Aschner, Khalaf F. Alsharif, Luciano Saso, Haroon Khan. "Marine Peptides in Prostate Cancer" Encyclopedia, https://encyclopedia.pub/entry/25616 (accessed July 22, 2026).
Ahmed, S., Alam, W., Jeandet, P., Aschner, M., Alsharif, K.F., Saso, L., & Khan, H. (2022, July 28). Marine Peptides in Prostate Cancer. In Encyclopedia. https://encyclopedia.pub/entry/25616
Ahmed, Salman, et al. "Marine Peptides in Prostate Cancer." Encyclopedia. Web. 28 July, 2022.
Copy Citation
Prostate cancer (PCa) is the leading cause of cancer death in men, and its treatment is commonly associated with severe adverse effects. Anticancer peptides are less toxic to normal cells and provide an efficacious treatment approach via multiple mechanisms, including altered cell viability, apoptosis, cell migration/invasion, suppression of angiogenesis and microtubule balance disturbances.
marine peptides
apoptosis
antimetastatic
antimitotic
1. Introduction
Prostate cancer (PCa) is one of the most often diagnosed cancers worldwide. It is the second leading source of cancer-related mortality in males, trailing behind only lung cancer, based on GLOBOCAN 2020 estimates [1]. Radiation and surgical procedures are used to treat this disease when it first appears and is localized. Despite a considerable increase in disease-free life following first surgical or radiation therapy, the illness recurs in more than 30% of patients. Androgen deprivation treatment (ADT) is the most often alternative for PCa treatment, because of the tumor’s requirement for male hormones for progression. This treatment is focused on pharmacological castration achieved via GnRH agonists alone or in conjunction with anti-androgens. However, despite an excellent initial response, most patients relapse within 2–3 years, and the tumor advances. Chemotherapeutic drugs, such as docetaxel, cabazitaxel, doxorubicin, abiraterone and enzalutamide, provide a few months of progression-free survival with highly toxic effects. Therefore, the development and refinement of unique anticancer drugs with minimal adverse side effects are required [2][3].
Natural products provide benefits over synthetic compounds because of their broader variety of targets, larger structural diversity and low toxicity against PCa [4][5]. Many bioactive substances found in the marine ecosystem have potential applications in the treatment of human diseases including cancer. A large number of novel sea-based biological compounds have been derived from corals, sponges, tunicates, bacteria, fungi, micro- and macroalgae, and other marine micro- and macro-organisms. Treatments have been impacted by marine-based medicines, with anticancer treatments made available with several marine chemical compounds. Indeed, clinical approval has been granted for several such treatments, including belantamabmafodotin, cytarabine, enfortumabvedotin, brentuximab vedotin, eribulin mesylate, lurbinectedin, fludarabine phosphate (prodrug of ara-A), nelarabine (prodrug of ara-G), polatuzumabvedotin, trabectedin, vidarabine, and plitidepsin [6][7][8][9]. Improvement in the physiological state of cancer patients depends on the introduction of natural strategies for clinical treatment. Due to their small size, ease of synthesis, capacity to cross cell membranes, low drug–drug interactions, precision targeting, and reduced side responses, marine peptides have also spurred attention in the development of anticancer medicines. The downsides of anticancer peptides include their short half-life, low bioavailability, poor pharmacokinetics, and protease sensitivity [10][11][12]. A schematic representation of the pathophysiology of prostate cancer is shown in Figure 1.
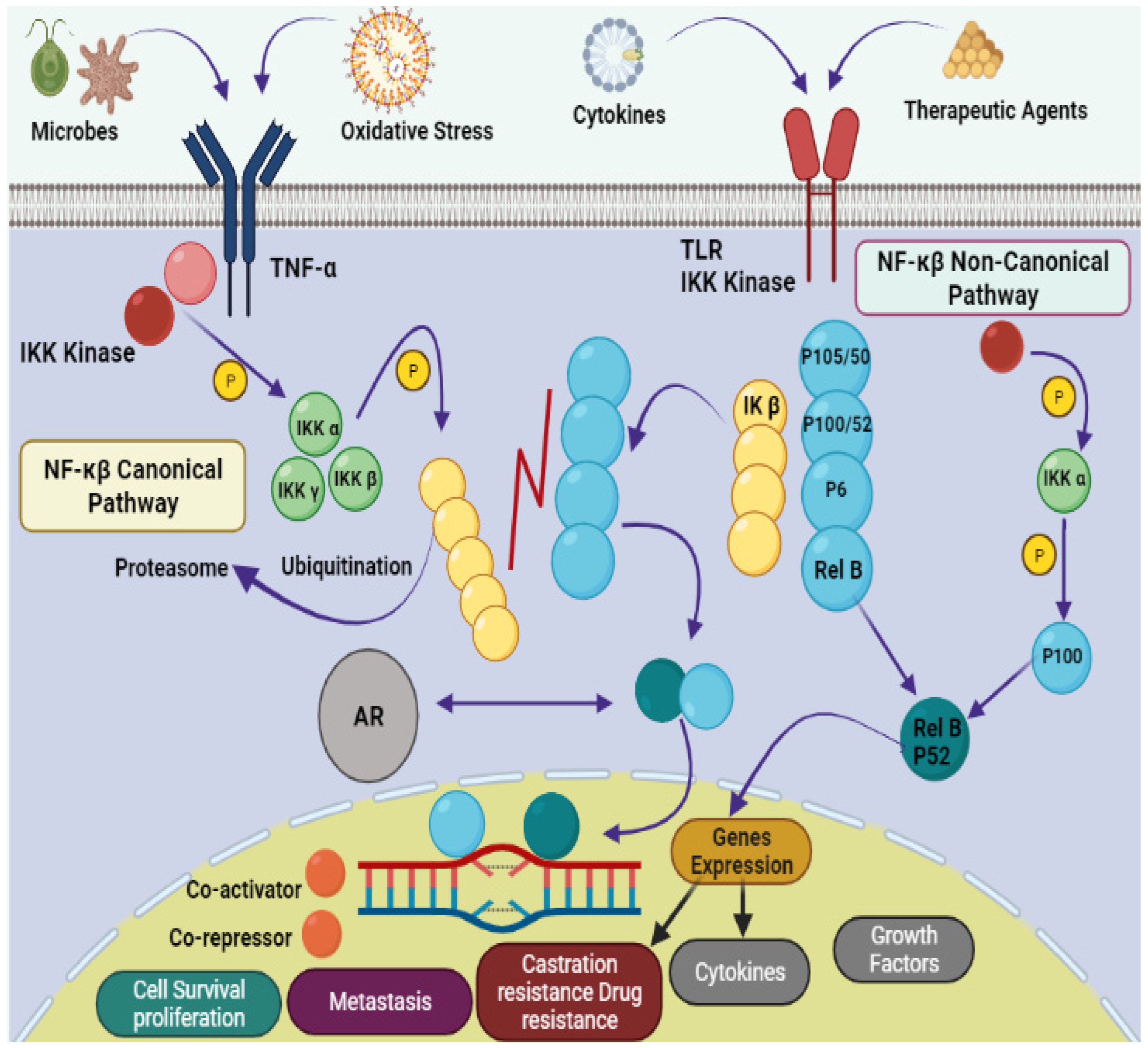
Figure 1. Pathophysiology of prostate cancer via NF-κB canonical and non-canonical pathways. The leading dimers of NF-κB are P50-P65, which activate the transcription process in canonical pathways. Different subunits like Toll-like receptor (TLR); tumor necrosis factor receptor (TNF-R); Inhibitor of NF-κB (IκB); IκB kinase; NF-κB-inducing kinase (NIK); mitogen-activated protein kinase (MAP); androgen receptor (AR); bone marrow-derived cell (BMDC) and major histocompatibility complex (MHC) are also involved in the pathology of prostate cancer.
Presently, more than 60% of clinically accessible anticancer medications have been derived from natural sources. Peptides are categorized depending on their ability to induce antioxidative mechanisms, cytotoxicity, apoptosis, inhibit cell proliferation, migration, and angiogenesis, microtubule-destabilization, or undiscovered mechanisms in diverse malignant cell lines, relating to clinical research for cancer treatment evaluation [12][13]. Anticancer peptides from algae, ascidians, bacteria, fungi, cyanobacteria, mollusks, sponges, and protein hydrolysates from clam, coral and fish, have been isolated. Linear and cyclic peptides are the two types of marine peptides. A straight-chain of amino acids linked together by amide bonds forms a linear peptide (Figure 2) [7].
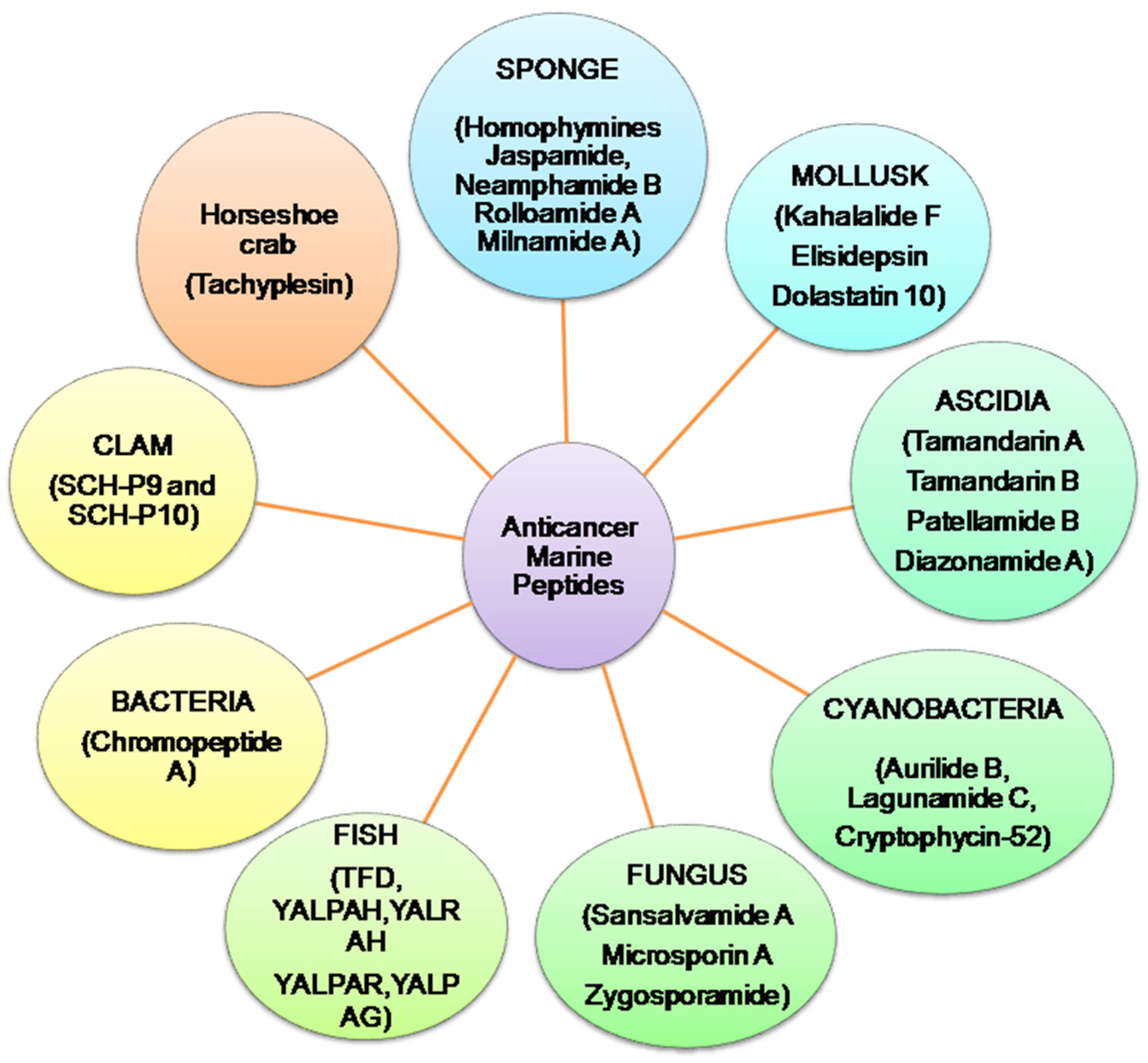
Figure 2. Summary of anticancer marine peptides isolated from different marine sources.
2. Mechanistic Insights
2.1. Apoptosis
One of the most critical mechanisms of cell death is apoptosis, and its failure is a severe barrier to cancer therapy. Cytochrome-c (cyt c) release leads to caspases activation and ensuing apoptosis [14]. C-phycocyanin from cyanobacteria [15] and AAP-H from sea anemone [16] have shown apoptotic efficacy by initiating cyt c release in DU145 and LNCaP cells. Caspases are the main executors of apoptosis, which are activated after proteolytic cleavage. Initiator caspases that include caspase-8, -9 and -10 initiate a regulated and programmed cell death cascade to trigger downstream caspases-3, -6, and -7 expression [17].
PI3K/AKT pathways play a significant role in regulating cell cycle and survival. AKT inhibitors attenuate the degree of BAK, BAX, and BAD phosphorylation, cause cyt c release, and activate casp-9 [18]. Decreased PI3K/AKT and ErbB3 levels are involved in cell cycle arrest as well as BAX and BAK activation [19]. PI3K/AKT and ErbB3 deficiency in PC-3 and DU145 have been noted upon treatment with Kahalalide F [20][21]. Elisidepsin or Irvalec (a Kahalalide F synthetic derivative) have been shown to inhibit PI3K/AKT and deplete ErbB3 in PC-3 and DU145 cells [22][23]. Furthermore, elisidepsin causes cellular swelling, plasma membrane rupture, and loss of intracellular contents, as well as necrotic cell death in PC-3 and 22RV1 at IC50s of 0.6M and 0.3M, respectively [24]. p38 mitogen-activated protein kinases (MAPKs) and Jun N-terminal kinases (JNKs) are activated by microtubule inhibitors, suggesting this may represent a general stress response to microtubule dysfunction. Cyt c release is induced by JNK and p38 MAPK activation, which, in turn, triggers caspase cascades. Activation of JNK and ERK induces mitochondrial-related apoptosis via JNK signaling and S phase cell cycle arrest via ERK signaling [25][26]. Cryptophycin-52 induces apoptosis in DU145 and LNCaP cells via caspases-3, -7; JNK, p38 MAPK and ERK activation, increasing BAX and decreasing Bcl2 and Bcl-xL expression (Figure 3) [27].
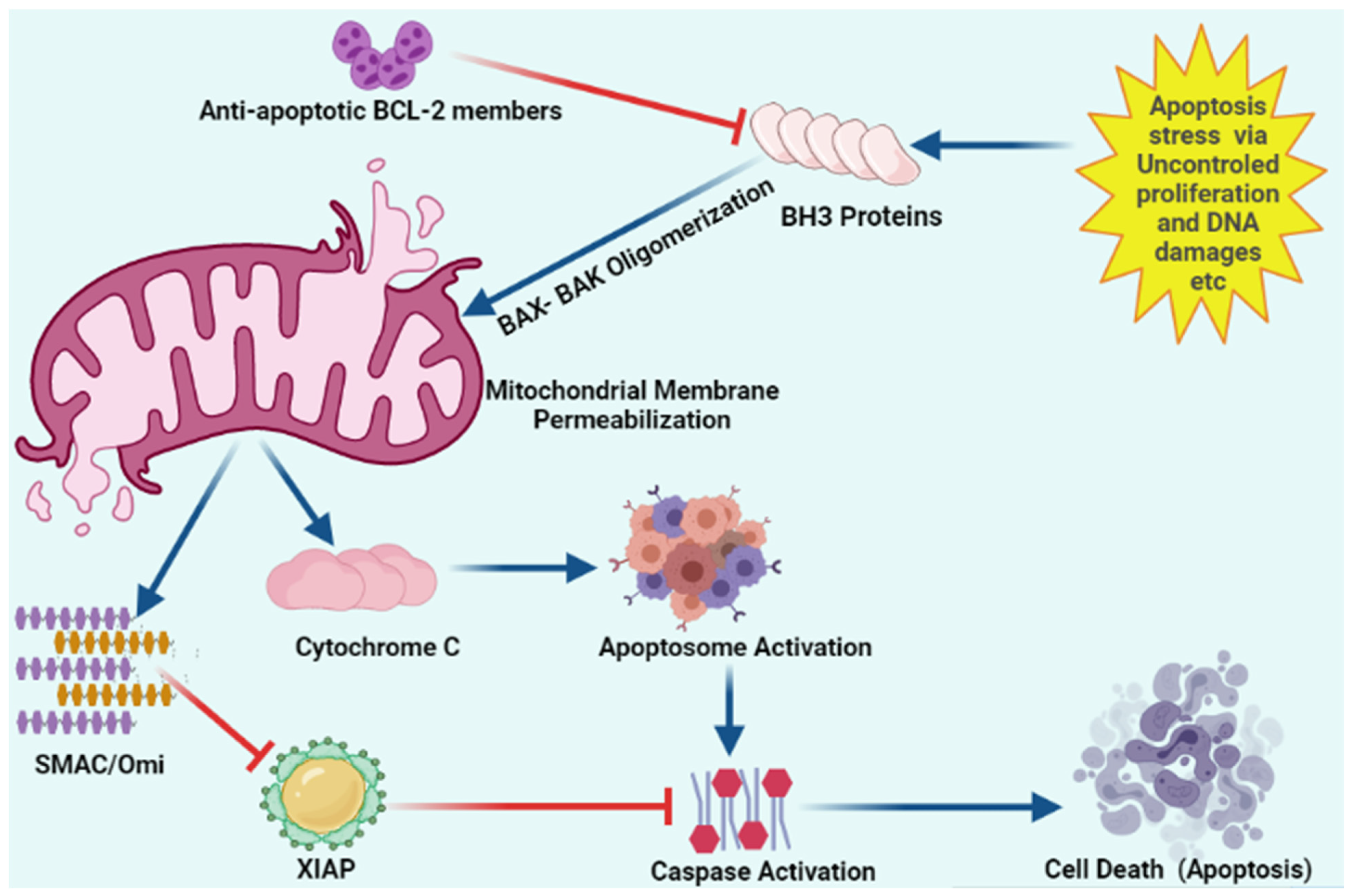
Figure 3. Schematic representation of intracellular apoptosis pathway.
Apoptosis stress involves mitochondrial outer membrane permeabilization via uncontrolled BH3 only proteins. BH-3 only proteins lead to oligomerization of BAK/BAX multimers. These BAK/BAX multimers within the outer membrane of the mitochondria form pores that allow cytochrome C release. Released cytochrome C interacts with Apaf-1 and pro-caspase-9 to form the apoptosome. Upon release, mitochondria-derived activator of caspase (SMAC) Cytochrome C and Omi activate apoptosome from procaspase-9 and cytochrome C. Caspases upon activation results in the cleavage of cellular proteins that leads to apoptosis. “Activation” is represented by blue arrows, whereas red T-bars show “inhibition”.
2.2. Antimitotic Effect
Antimitotic drugs function by stabilizing and destabilizing microtubule dynamics, as well as shifting the balance between tubulin polymerization and depolymerization. The majority of these drugs act via G2/M phase arrest [28]. Microtubules provide a variety of critical cellular activities, including chromosomal segregation, cell shape preservation, transport, motility, and organelle distribution. Microtubules, the key components of the mitotic spindle, play an important role in cell division. Microtubular dynamic disruption arrests the cell cycle at the metaphase–anaphase transition leading to cell death [29]. Hemiasterlin and its analogue HTI-286 depolymerize microtubules by disrupting microtubular dynamics in LNCaP, C4-2, PC-3, PC-3dR cell lines with IC50s in the range of 0.65–4.6 nM. The same effect has been noted in PC3-MM2, PC-3 and PC-3dR xenografts at 1–1.5 mg/kg i.v. [30][31]. Dolastatin 10 (IC50:0.5 nM) inhibits microtubule assembly in DU145 cells [32]. Analogous behavior has been observed for Diazonamide A in PC-3 cells with IC50 of 2.3 nM [33]. Cryptophycin-52 (LY355703), a synthetic cryptophycin, inhibits DU145 and LNCaP cell growth during mitosis by depolymerizing spindle microtubules and alters chromosomal organization [27]. By attaching to the microtubules, microtubule-stabilizing drugs promote microtubule polymerization and target the cytoskeleton and spindle apparatus of tumor cells, leading to mitotic interruption [29]. Aurilide B has been shown to cause microtubular destabilization in PC-3 and DU145 carcinoma cell lines with GI50 < 10 nM [34].
2.3. Antimetastatic Activity
Non-caspase proteases (elastase, trypsin and chymotrypsin) are critical regulators of PCa progression. The PCa metastatic cascade is characterized by a defined chain of steps, beginning with neoangiogenesis or lymphangiogenesis, culminating in the loss of tumor cell adhesion, local invasion of host stroma, and tumor cell escape into the vasculature or lymphatics, and eventually dissemination, extravasation, and colonization of specific metastatic sites. Proteases secrete angiogenic factors, cell adhesion molecules, breakdown basement membranes, induce epithelial–mesenchymal transition, participate in extravasation, and are necessary for metastatic site colonization. Several proteases are increased in tumor cells, and have specific roles in facilitating various phases of this cascade [35][36]. Trypsin plays a tumorigenic role in PCa and suppressing trypsin/mesotrypsin activity may provide a new PCa therapeutic strategy. PC-3 cells originating from a grade IV prostate cancer bone metastases exhibit an extremely significant overexpression of PRSS3/mesotrypsin [37]. LNCaP human prostate cells have shown upregulation of chymotrypsin-like proteasomal activity, suggesting the involvement of chymotrypsin in PCa [38]. Elastase increases PCa proliferation, migration, invasion and has been used as a therapeutic target [39].
The cytoskeletal microfilament, actin, is required for cytokinesis, cell migration, and a host of other processes crucial for the stability of cancerous cells. Inhibiting actin polymerization slows the growth of metastatic neoplastic cells by causing the breakdown of microfilaments, which, in turn, reduces cell motility [40]. Jaspamide, a cyclicdepsipeptide from sponge (Jaspis johnstoni), has shown antiproliferative activity against DU145, LNCaP, and PC-3 with IC50s of 0.8, 0.07 and 0.3µM by actin filament disruption. The same peptide has shown anticancer activity in a DU-145 xenograft [40].
Voltage-gated sodium channels (VGSC) are considered to have a role in cancer cell invasion and metastasis. In PCa, VGSC overexpression is crucial for cell movement and invasiveness [41][42][43]. Palmyramide A, a cyclic depsipeptide with an IC50 of 17.2 μM, and hermitamides A and B (lipopeptides) with IC50s of 1 μM have been shown to block sodium channels via VGSC inhibition [44][45].
2.4. Antiangiogenic Effect
Angiogenesis is crucial in the development of cancer [45]. VEGF is produced in cancer cells and is required for angiogenesis. During low oxygen (hypoxia) periods, Mucin 1 (MUC1) increases HIF-1α to stimulate tumor development and angiogenesis. Its overexpression restricts apoptosis via upregulating Bcl-xL and inactivating BAD protein. A decline in VEGF expression level is associated with MUC1 silencing, establishing that MUC1 downregulation has an anti-angiogenic impact [46][47][48]. TFD and SIO peptides from fish inhibit PC-3 and DU145 cell migration by decreasing VEGFR1 and MUC1 protein expression [49][50][51].
2.5. Cell Cycle Arrest
Cell cycle arrest limits cell viability and is related to apoptosis [52]. The two main regulators of G2/M transition/progression are cdc2 and cell division cycle-25C (cdc25C). Multiple signaling pathways influence their regulation in the cell cycle, and are linked to carcinogenesis and tumor formation. cdc2 and cdc25C have been shown to enhance mitotic cell G2/M transition by dephosphorylating cyclin-dependent kinase-1 (CDK1) and activating the cyclin B1/CDK1 complex. Their downregulation causes G2/M cell cycle arrest via p53-mediated signal transduction [53]. cdc2 and cdc25C are highly expressed in PCa [54]. Chromopeptide A promotes G2/M phase arrest in PCa cells by suppressing cdc2 and cdc25C phosphorylation [55]. Similarly, cryptophycin-52 induces G2/M phase arrest in DU145 and LNCaP cells [27].
2.6. p53 Upregulation
The functional tumor protein p53 (p53) protein takes part in apoptosis initiation via BAK, BAX increment and Bcl2, Bcl-xL decrement. Cells also arrest in the G1 and G2/M stages when p53 is activated. Low p53 level has been detected in PCa [56][57][58]. Cryptophycin-52 [27], chromopeptide A [55] and sepia ink peptides [49][50][59] induce p53 upregulation in PC-3, DU145 and LNCaP cells and hence regulate p53-dependent apoptosis and cell cycle arrest.
2.7. Stimulation of Histone Hyperacetylation
Histone deacetylases (HDACs) are widely produced and over-activated in PCa. Stimulation of histone hyperacetylation in tumor through cellular HDAC inhibition results in G2/M phase arrest, apoptosis, activates p53, DNA-damage response and inhibition of metastasis and angiogenesis [60]. The chromopeptide A from marine-derived Chromobacterium sp. stimulates histone hyperacetylation by HDAC inhibition in PC-3, DU145, LNCaP cell lines and human PC-3 xenograft mouse model [55].
2.8. Mitochondrial Dysfunctions and Oxidative Damage
Reactive oxygen species (ROS) accumulation induces oxidative stress caused by mitochondrial abnormalities, and malignant cells require high ROS concentrations [61]. The most frequent type of DNA damage is DNA fragmentation, a direct consequence of oxidative stress [62]. Dolastatin 10 induces DNA damage in DU145 [32]. Similarly, Cryptophycin 52 and C-phycocyanin induce DNA damage in DU145 and LNCaP [15][27]. A schematic representation of the anticancer mechanisms of marine peptides is depicted in Figure 4 as under:
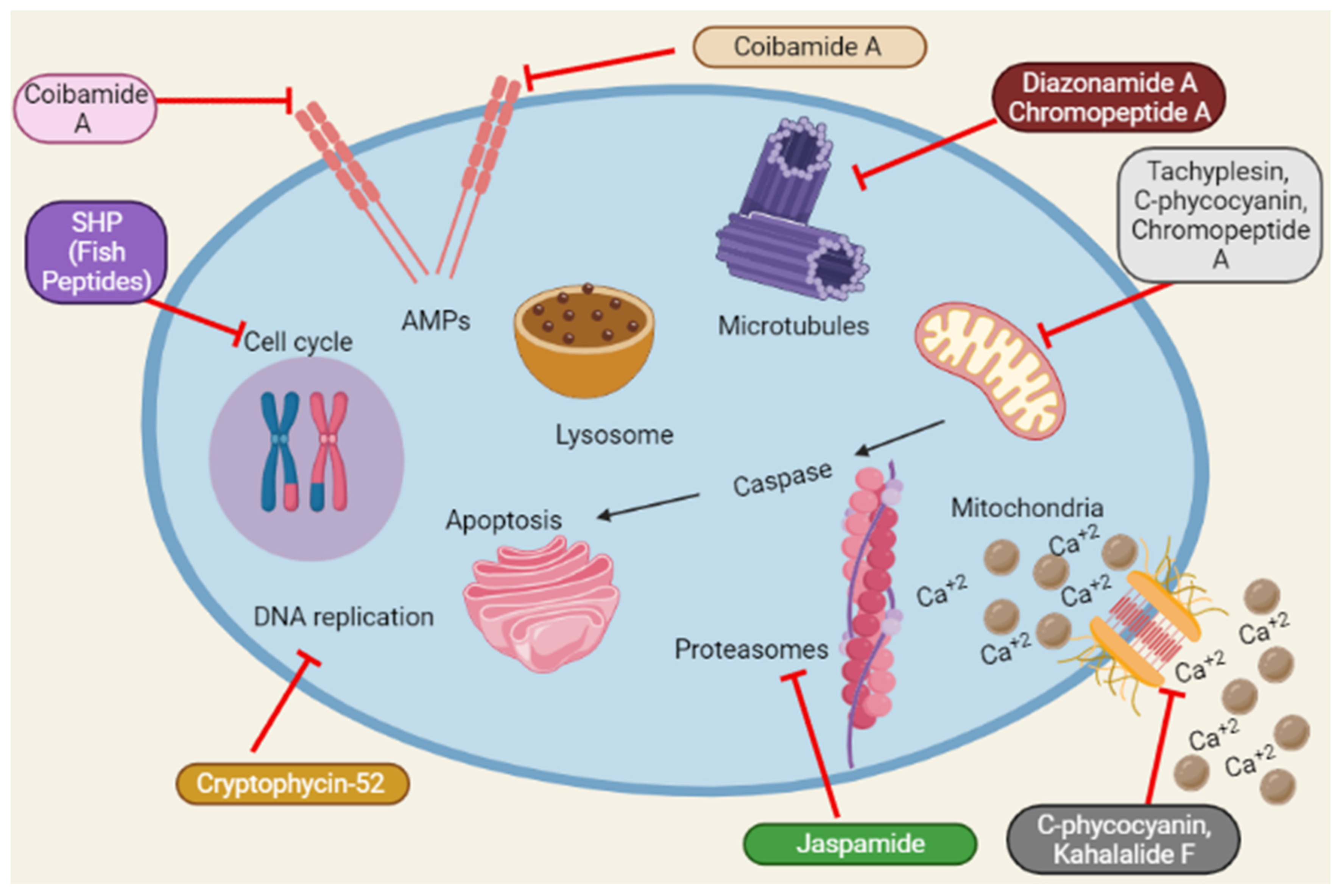
Figure 4. Summary of the schematic representation of the anticancer mechanisms of marine peptides at different cellular pathways. Marine peptides inhibit different pathways such as inhibition of cell cycle, Amps, caspase, Ca+2 influx, DNA replication, protein synthesis and lysosomal pathways.
2.9. Unidentified Mechanisms for Anticancer Activity
Geodiamolides D–F [30], homophymines A–E [63], milnamides A–G [30], neamphamides B–D [64], rolloamide A [65], yaku’amides A and B [66] from sponges; lagunamide C [67], coibamide A [68], laxaphycin B [69] from cyanobacteria exhibit strong cytotoxicity in several PCa cells, although the specific targets are yet unknown. Patellamides B and F, ulithiacyclamide [70], trunkamide A [12][71], tamandarins A-B [72][73] from ascidia also elicit anti-PCa activity via unrevealed process. Microsporin A [74], sansalvamide A [75] and zygosporamide [76] from marine derived fungus; YALPAH from fish [77] possess anti-PCa properties via an unknown mechanism.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249.
- Rebello, R.J.; Oing, C.; Knudsen, K.E.; Loeb, S.; Johnson, D.C.; Reiter, R.E.; Gillessen, S.; Van der Kwast, T.; Bristow, R.G. Prostate cancer. Nat. Rev. Dis. Primers 2021, 7, 9.
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89.
- Mokbel, K.; Wazir, U.; Mokbel, K. Chemoprevention of Prostate Cancer by Natural Agents: Evidence from Molecular and Epidemiological Studies. Anticancer. Res. 2019, 39, 5231.
- Fontana, F.; Raimondi, M.; Marzagalli, M.; Di Domizio, A.; Limonta, P. Natural Compounds in Prostate Cancer Prevention and Treatment: Mechanisms of Action and Molecular Targets. Cells 2020, 9, 460.
- Alves, C.; Diederich, M. Marine Natural Products as Anticancer Agents. Mar. Drugs 2021, 19, 447.
- Barreca, M.; Spanò, V.; Montalbano, A.; Cueto, M.; Marrero, A.R.D.; Deniz, I.; Erdoğan, A.; Bilela, L.L.; Moulin, C.; Taffin-De-Givenchy, E.; et al. Marine Anticancer Agents: An Overview with a Particular Focus on Their Chemical Classes. Mar. Drugs 2020, 18, 619.
- Ahmed, S.; Mirzaei, H.; Aschner, M.; Khan, A.; Al-Harrasi, A.; Khan, H. Marine peptides in breast cancer: Therapeutic and mechanistic understanding. Biomed. Pharmacother. 2021, 142, 112038.
- Ahmed, S.; Hasan, M.M.; Aschner, M.; Mirzaei, H.; Alam, W.; Shah, S.M.M.; Khan, H. Therapeutic potential of marine peptides in glioblastoma: Mechanistic insights. Cell. Signal. 2021, 87, 110142.
- Kang, H.K.; Choi, M.-C.; Seo, C.H.; Park, Y. Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides. Int. J. Mol. Sci. 2018, 19, 919.
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application (Review). Int. J. Oncol. 2020, 57, 678–696.
- Zhang, Q.-T.; Liu, Z.-D.; Wang, Z.; Wang, T.; Wang, N.; Wang, N.; Zhang, B.; Zhao, Y.-F. Recent Advances in Small Peptides of Marine Origin in Cancer Therapy. Mar. Drugs 2021, 19, 115.
- Ucak, I.; Afreen, M.; Montesano, D.; Carrillo, C.; Tomasevic, I.; Simal-Gandara, J.; Barba, F. Functional and Bioactive Properties of Peptides Derived from Marine Side Streams. Mar. Drugs 2021, 19, 71.
- Pfeffer, C.M.; Singh, A.T. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448.
- Gantar, M.; Dhandayuthapani, S.; Rathinavelu, A. Phycocyanin Induces Apoptosis and Enhances the Effect of Topotecan on Prostate Cell Line LNCaP. J. Med. Food 2012, 15, 1091–1095.
- Wu, Z.-Z.; Ding, G.F.; Huang, F.F.; Yang, Z.S.; Yu, F.M.; Tang, Y.P.; Jia, Y.L.; Zheng, Y.Y.; Chen, R. Anticancer Activity of Anthopleura anjunae Oligopeptides in Prostate Cancer DU-145 Cells. Mar. Drugs 2018, 16, 125.
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672.
- Jeong, S.-J.; Dasgupta, A.; Jung, K.-J.; Um, J.-H.; Burke, A.; Park, H.U.; Brady, J.N. PI3K/AKT inhibition induces caspase-dependent apoptosis in HTLV-1-transformed cells. Virology 2008, 370, 264–272.
- Lee, H.; Chin, H.; Kim, K.; Lee, D. ERBB3 knockdown induces cell cycle arrest and activation of Bak and Bax-dependent apoptosis in colon cancer cells. Oncotarget 2014, 5, 5138–5152.
- Suárez, Y.; González, L.; Cuadrado, A.; Berciano, M.; Lafarga, M.; Muñoz, A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol. Cancer Ther. 2003, 2, 863.
- Janmaat, M.L.; Rodriguez, J.A.; Jimeno, J.; Kruyt, F.A.E.; Giaccone, G. Kahalalide F Induces Necrosis-Like Cell Death that Involves Depletion of ErbB3 and Inhibition of Akt Signaling. Mol. Pharmacol. 2005, 68, 502.
- Serova, M.; de Gramont, A.; Bieche, I.; Riveiro, M.E.; Galmarini, C.M.; Aracil, M.; Jimeno, J.; Faivre, S.; Raymond, E. Predictive factors of sensitivity to elisidepsin, a novel Kahalalide F-derived marine compound. Mar. Drugs 2013, 11, 944–959.
- Wang, E.; Sorolla, M.A.; Krishnan, P.D.G.; Sorolla, A. From Seabed to Bedside: A Review on Promising Marine Anticancer Compounds. Biomolecules 2020, 10, 248.
- Molina-Guijarro, J.M.; Macías, Á.; García, C.; Muñoz, E.; García-Fernandez, L.F.; David, M.; Núñez, L.; Martínez-Leal, J.F.; Moneo, V.; Cuevas, C.; et al. Irvalec inserts into the plasma membrane causing rapid loss of integrity and necrotic cell death in tumor cells. PLoS ONE 2011, 6, e19042.
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346.
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366.
- Drew, L.; Fine, R.L.; Do, T.N.; Douglas, G.P.; Petrylak, D.P. The Novel Antimicrotubule Agent Cryptophycin 52 (LY355703) Induces Apoptosis via Multiple Pathways in Human Prostate Cancer Cells. Clin. Cancer Res. 2002, 8, 3922.
- van Vuuren, R.J.; Visagie, M.H.; Theron, A.E.; Joubert, A.M. Antimitotic drugs in the treatment of cancer. Cancer Chemother. Pharmacol. 2015, 76, 1101–1112.
- Fanale, D.; Bronte, G.; Passiglia, F.; Calò, V.; Castiglia, C.; Di Piazza, F.; Barraco, N.; Cangemi, A.; Catarella, M.T.; Insalaco, L.; et al. Stabilizing versus destabilizing the microtubules: A double-edge sword for an effective cancer treatment option? Anal. Cell. Pathol. 2015, 2015, 690916.
- Tran, T.D.; Pham, N.B.; Fechner, G.A.; Hooper, J.N.A.; Quinn, R.J. Potent cytotoxic peptides from the Australian marine sponge Pipestela candelabra. Mar. Drugs 2014, 12, 3399–3415.
- Alves, C.; Silva, J.; Pinteus, S.; Gaspar, H.; Alpoim, M.C.; Botana, L.M.; Pedrosa, R. From Marine Origin to Therapeutics: The Antitumor Potential of Marine Algae-Derived Compounds. Front. Pharmacol. 2018, 9, 777.
- Turner, T.; Jackson, W.H.; Pettit, G.R.; Wells, A.; Kraft, A.S. Treatment of human prostate cancer cells with dolastatin 10, a peptide isolated from a marine shell-less mollusc. Prostate 1998, 34, 175–181.
- Cruz-Monserrate, Z.; Vervoort, H.C.; Bai, R.; Newman, D.J.; Howell, S.B.; Los, G.; Mullaney, J.T.; Williams, M.D.; Pettit, G.R.; Fenical, W.; et al. Diazonamide A and a Synthetic Structural Analog: Disruptive Effects on Mitosis and Cellular Microtubules and Analysis of Their Interactions with Tubulin. Mol. Pharmacol. 2003, 63, 1273.
- Han, B.; Gross, H.; Goeger, D.E.; Mooberry, S.L.; Gerwick, W.H. Aurilides B and C, Cancer Cell Toxins from a Papua New Guinea Collection of the Marine Cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2006, 69, 572–575.
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011.
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691.
- Hockla, A.; Miller, E.; Salameh, M.D.A.; Copland, J.A.; Radisky, D.C.; Radisky, E.S. PRSS3/mesotrypsin is a therapeutic target for metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 1555–1566.
- Wei, X.; Zeng, W.; Xie, K.; Diao, P.; Tang, P. Potential use of chymotrypsin-like proteasomal activity as a biomarker for prostate cancer. Oncol. Lett. 2018, 15, 5149–5154.
- Lerman, I. Neutrophil Elastase and SERPINB1 Are Critical Regulators of Prostate Cancer Progression; University of Rochester: Ann Arbor, MI, USA, 2018; p. 159.
- Jiang, X.; Qin, Y.; Kun, L.; Zhou, Y. The Significant Role of the Microfilament System in Tumors. Front. Oncol. 2021, 11, 620390.
- Yildirim, S.; Altun, S.; Gumushan, H.; Patel, A.; Djamgoz, M.B. Voltage-gated sodium channel activity promotes prostate cancer metastasis in vivo. Cancer Lett. 2012, 323, 58–61.
- Angus, M.; Ruben, P. Voltage gated sodium channels in cancer and their potential mechanisms of action. Channels 2019, 13, 400–409.
- Mao, W.; Zhang, J.; Körner, H.; Jiang, Y.; Ying, S. The Emerging Role of Voltage-Gated Sodium Channels in Tumor Biology. Front. Oncol. 2019, 9, 124.
- De Oliveira, E.O.; Graf, K.M.; Patel, M.K.; Baheti, A.; Kong, H.-S.; MacArthur, L.H.; Dakshanamurthy, S.; Wang, K.; Brown, M.L.; Paige, M. Synthesis and evaluation of hermitamides A and B as human voltage-gated sodium channel blockers. Bioorganic Med. Chem. 2011, 19, 4322–4329.
- Taniguchi, M.; Nunnery, J.K.; Engene, N.; Esquenazi, E.; Byrum, T.; Dorrestein, P.C.; Gerwick, W.H. Palmyramide A, a Cyclic Depsipeptide from a Palmyra Atoll Collection of the Marine Cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2010, 73, 393–398.
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105.
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 Signaling and its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int. J. Mol. Sci. 2020, 21, 1388.
- Khodabakhsh, F.; Merikhian, P.; Eisavand, M.R.; Farahmand, L. Crosstalk between MUC1 and VEGF in angiogenesis and metastasis: A review highlighting roles of the MUC1 with an emphasis on metastatic and angiogenic signaling. Cancer Cell Int. 2021, 21, 200.
- Ding, G.-F.; Huang, F.F.; Yang, Z.-S.; Yu, D.; Yang, Y.F. Anticancer Activity of an Oligopeptide Isolated from Hydrolysates of Sepia Ink. Chin. J. Nat. Med. 2011, 9, 151–155.
- Jing, Y.; Yang, Z.; Huang, F. Mechanism of Sepia ink polypeptide-induced apoptosis in DU-145 prostate cancer cells. Mod. Food Sci. Technol. 2014, 30, 1–6.
- Guha, P.; Kaptan, E.; Bandyopadhyaya, G.; Kaczanowska, S.; Davila, E.; Thompson, K.; Martin, S.S.; Kalvakolanu, D.V.; Vasta, G.R.; Ahmed, H. Cod glycopeptide with picomolar affinity to galectin-3 suppresses T-cell apoptosis and prostate cancer metastasis. Proc. Natl. Acad. Sci. 2013, 110, 5052–5057.
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. CR 2016, 35, 153.
- Liu, K.; Zheng, M.; Lu, R.; Du, J.; Zhao, Q.; Li, Z.; Li, Y.; Zhang, S. The role of CDC25C in cell cycle regulation and clinical cancer therapy: A systematic review. Cancer Cell Int. 2020, 20, 213.
- Ozen, M.; Ittmann, M. Increased Expression and Activity of CDC25C Phosphatase and an Alternatively Spliced Variant in Prostate Cancer. Clin. Cancer Res. 2005, 11, 4701.
- Sun, J.-Y.; Wang, J.-D.; Wang, X.; Liu, H.-C.; Zhang, M.-M.; Liu, Y.-C.; Zhang, C.-H.; Su, Y.; Shen, Y.-Y.; Guo, Y.-W.; et al. Marine-derived chromopeptide A, a novel class I HDAC inhibitor, suppresses human prostate cancer cell proliferation and migration. Acta Pharmacol. Sin. 2017, 38, 551–560.
- Wan, J.; Zhang, J.; Zhang, J. Expression of p53 and its mechanism in prostate cancer. Oncol. Lett. 2018, 16, 378–382.
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; Murphy, M.E. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521.
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104.
- Huang, F.; Jing, Y.; Ding, G.; Yang, Z. Isolation and purification of novel peptides derived from Sepia ink: Effects on apoptosis of prostate cancer cell PC-3. Mol. Med. Rep. 2017, 16, 4222–4228.
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219.
- Zampella, A.; Sepe, V.; Bellotta, F.; Luciano, P.; D’Auria, M.V.; Cresteil, T.; Debitus, C.; Petek, S.; Poupat, C.; Ahond, A. Homophymines B–E and A1–E1, a family of bioactive cyclodepsipeptides from the sponge Homophymia sp. Org. Biomol. Chem. 2009, 7, 4037–4044.
- Tran, T.D.; Pham, N.B.; Fechner, G.; Zencak, D.; Vu, H.T.; Hooper, J.N.; Quinn, R.J. Cytotoxic Cyclic Depsipeptides from the Australian Marine Sponge Neamphius huxleyi. J. Nat. Prod. 2012, 75, 2200–2208.
- Williams, D.E.; Yu, K.; Behrisch, H.W.; Van Soest, R.; Andersen, R.J. Rolloamides A and B, Cytotoxic Cyclic Heptapeptides Isolated from the Caribbean Marine Sponge Eurypon laughlini. J. Nat. Prod. 2009, 72, 1253–1257.
- Ueoka, R.; Ise, Y.; Ohtsuka, S.; Okada, S.; Yamori, T.; Matsunaga, S. Yaku’amides A and B, Cytotoxic Linear Peptides Rich in Dehydroamino Acids from the Marine Sponge Ceratopsions. J. Am. Chem. Soc. 2010, 132, 17692–17694.
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Rottmann, M.; Chan, K.P.; Chen, D.Y.K.; Tan, L.T. Lagunamide C, a cytotoxic cyclodepsipeptide from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 2011, 72, 2369–2375.
- Medina, R.A.; Goeger, D.E.; Hills, P.; Mooberry, S.L.; Huang, N.; Romero, L.I.; Ortega-Barría, E.; Gerwick, W.H.; McPhail, K.L. Coibamide A, a potent antiproliferative cyclic depsipeptide from the Panamanian marine cyanobacterium Leptolyngbya sp. J. Am. Chem. Soc. 2008, 130, 6324–6325.
- Bonnard, I.; Rolland, M.; Salmon, J.-M.; Debiton, E.; Barthomeuf, C.; Banaigs, B. Total Structure and Inhibition of Tumor Cell Proliferation of Laxaphycins. J. Med. Chem. 2007, 50, 1266–1279.
- Rashid, M.A.; Gustafson, K.R.; Ii, J.H.C.; Boyd, M.R. Patellamide F, a New Cytotoxic Cyclic Peptide from the Colonial Ascidian Lissoclinum patella. J. Nat. Prod. 1995, 58, 594–597.
- McKeever, B.; Pattenden, G. Total synthesis of trunkamide A, a novel thiazoline-based prenylated cyclopeptide metabolite from Lissoclinum sp. Tetrahedron 2003, 59, 2713–2727.
- Vervoort, H.; Fenical, W.; Epifanio, R.D.A. Tamandarins A and B: New Cytotoxic Depsipeptides from a Brazilian Ascidian of the Family Didemnidae. J. Org. Chem. 2000, 65, 782–792.
- Liang, B.; Richard, D.J.; Portonovo, P.S.; Joullie, M.M. Total Syntheses and Biological Investigations of Tamandarins A and B and TamandarinA Analogs. J. Am. Chem. Soc. 2001, 123, 4469–4474.
- Gu, W.; Cueto, M.; Jensen, P.R.; Fenical, W.; Silverman, R.B. Microsporins A and B: New histone deacetylase inhibitors from the marine-derived fungus Microsporum cf. gypseum and the solid-phase synthesis of microsporin A. Tetrahedron 2007, 63, 6535–6541.
- Liu, S.; Gu, W.; Lo, D.; Ding, X.-Z.; Ujiki, M.; Adrian, T.E.; Soff, G.A.; Silverman, R.B. N-Methylsansalvamide A Peptide Analogues. Potent New Antitumor Agents. J. Med. Chem. 2005, 48, 3630–3638.
- Oh, D.-C.; Jensen, P.R.; Fenical, W. Zygosporamide, a cytotoxic cyclic depsipeptide from the marine-derived fungus Zygosporium masonii. Tetrahedron Lett. 2006, 47, 8625–8628.
- Song, R.; Wei, R.B.; Luo, H.Y.; Yang, Z.S. Isolation and identification of an antiproliferative peptide derived from heated products of peptic hydrolysates of half-fin anchovy (Setipinnataty). J. Funct. Foods 2014, 10, 104–111.
More
Information
Subjects:
Others
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Entry Collection:
Peptides for Health Benefits
Revisions:
2 times
(View History)
Update Date:
29 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No