Successful whole genome amplification (WGA) is a cornerstone of contemporary preimplantation genetic testing (PGT). Choosing the most suitable WGA technique for PGT can be particularly challenging.
1. Introduction
Applications of whole genome amplification (WGA) are widespread in clinical and scientific practices dealing with a limited amount of starting genetic material that is often of poor quality. Such practices include human identification in forensic and archeological investigations, microbiological studies, and prenatal and preimplantation genetic testing (PGT). PGT is clinically performed to exclude chromosomal or genetic pathology in the embryo with the aim of increasing embryo implantation rates or conceiving a healthy child in a family with a certain monogenic disease. Embryo genetic testing for monogenic disorders could only be realized after the pioneering of polymerase chain reaction (PCR) in 1985, and was first performed in 1989
[1]. With time, PGT has undergone significant methodological and approach changes in terms of clinical indications, embryological toolkit (manual versus laser-assisted biopsy and freezing versus vitrification), analysed material (blastomeres, polar bodies, trophectoderm cells and even morula cells), and, most importantly, downstream genetic applications, to improve conventional PCR (nested PCR, multiplex PCR, and fluorescent PCR).
In 1992, two well-known WGA technologies were developed–primer extension preamplification PCR (PEP-PCR) and degenerate oligonucleotide primer PCR (DOP-PCR)
[2][3]. WGA involves the use of random or partially random primers with the aim of efficiently replicating limited amounts of a target genome in a sequence-independent manner. Due to the nature of the procedure, any WGA is prone to drawbacks such as incomplete genomic coverage and amplification bias. Notwithstanding these shortcomings, WGA advancement was a turning point for PGT. Access to an almost endless supply of otherwise minute amounts of amplified DNA of a single or a few cells allowed for multilocus analysis, leading to a more versatile and safer PGT practice.
The majority of PGT cycles are performed to exclude the main cause of failed embryo implantation–chromosomal aneuploidies
[4]—termed PGT-A. The next most common reason to perform PGT is structural chromosomal rearrangements in one of the parents’ karyotypes, most often Robertsonian or reciprocal translocations. Classical PGT for structural rearrangements, termed PGT-SR, can only distinguish between balanced and unbalanced embryos, without an option to identify carrier embryos. Being straightforward and not requiring any specific preparation, prerequisites of PGT-A and classical PGT-SR are successful embryo biopsy and WGA. This is because aneuploidy or, in a broader concept, copy number variation (CNV) screening is performed in a genome-wide manner commonly using array comparative genomic hybridization (aCGH) or massively parallel sequencing (MPS), thus requiring substantial amounts of genetic material.
In contrast, embryo assessment for a particular single gene or monogenic disorder (PGT-M) can be particularly challenging and prone to several biological and technical drawbacks. These include crossover events adjacent to the locus of interest, consanguinity, de novo mutations (biological), decreased methodological resolution, DNA contamination, and allelic dropout (ADO), which is amplification failure affecting one or both of the parental alleles (technical)
[5]. Classical PGT-M requires customization in advance of the clinical test in order to construct family haplotypes—a practice that is time consuming and only useful for the particular gene/locus or sometimes only for the family being tested. Haplotyping is essential to track the transmission of a disease allele to an embryo and is commonly done through the assessment of polymorphic markers—short tandem repeats (STRs) or single nucleotide polymorphisms/variations (SNPs/SNVs)—adjacent to the mutation locus
[6][7]. Thus, being an inherently multifactor analysis, PGT-M also heavily relies on the merits of WGA.
As the first step of PGT-M and PGT-SR cycles frequently involves WGA, this allows for additional aneuploidy screening in the same biopsy sample—a practice termed multifactor PGT. The exclusion of chromosomal aneuploidies in PGT-M and PGT-SR cycles has been reported to be associated with improved pregnancy rates
[5][8]. As a consequence, it is being increasingly requested in clinics. However, running several methods in parallel complicates and prolongs the practical application of mainstream PGT. To overcome the obstacles associated with multifactor PGT performed in a parallel manner, application specialists are pursuing time-saving and simplified comprehensive PGT (cPGT) approaches, combining PGT-M, PGT-A, and PGT-SR in a single assay (
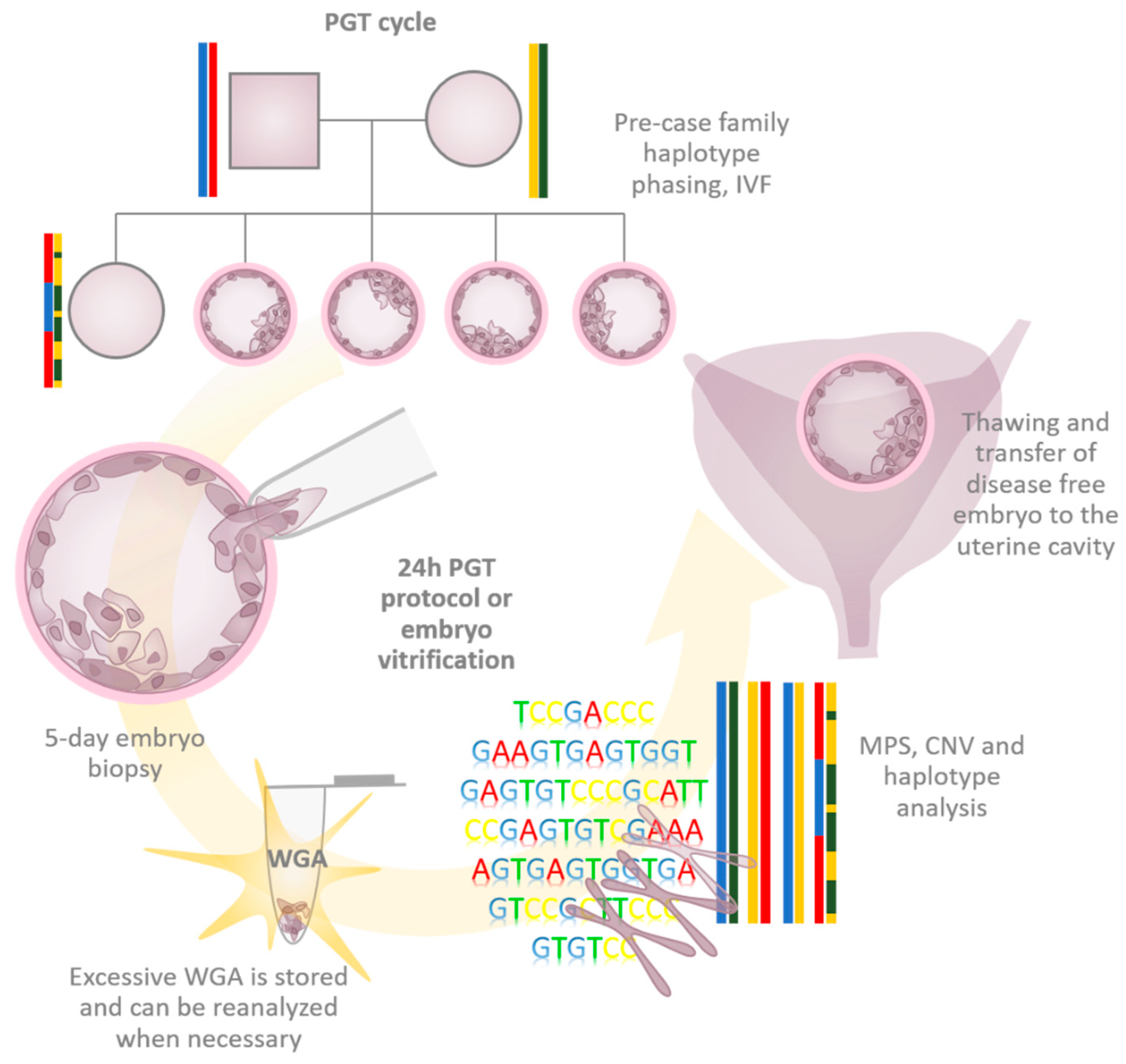
Figure 1) and universally suiting every case. In such a scenario, PGT-M is performed based on a genome-wide haplotyping approach requiring minimal or no custom preparation. Being clinically attractive, cPGT puts serious demands on the technical and molecular execution of the universal application and especially on WGA, the starting point of any embryo genetic testing in the era of genomics.
Figure 1. Clinical cycle of comprehensive preimplantation genetic testing. The process starts with family genetic testing in order to establish normal and disease-associated family haplotypes. Next, oocyte stimulation, retrieval, and fertilization are performed. Embryos surviving until day 5 undergo laser-assisted hatching and biopsy. Biopsy material consisting of a few trophectoderm cells is amplified using one of the WGA techniques. WGA material is further used to assess the embryonic genome, including haplotype, causative variant, aneuploidy, and CNV analyses. The disease-free embryo is subjected to embryo transfer to the uterine cavity. The whole cycle can be repeated within 24 h, with a day-6 embryo being transferred. If the genetic testing takes longer, then embryos are vitrified and thawed before being transferred. PGT—preimplantation genetic testing, WGA—whole-genome amplification, IVF—in vitro fertilization, MPS—massively parallel sequencing, CNV—copy number variation.
2. Requirements of WGA in PGT
Unlike any other application, PGT deals with very precious and sensitive material, namely the DNA of a developing embryo. Thus, any actions and manipulations of an embryo or its parts are associated with certain limitations and peculiarities. First, biopsy of an embryo needs to be done in the least traumatizing manner, with successful removal of as few cells as possible during the first attempt. The safest practice is now accepted to be trophectoderm biopsy of a day-5 developing embryo
[9][10].
In addition to the high demands placed on embryological personnel, they are also placed on the WGA performance itself. The most important requisite originally required from WGA for PGT—to provide an abundance of template DNA—is still as important today. One diploid cell contains just 6–7 ng of DNA, whereas WGA is expected to yield at least a few micrograms of the reaction product to suffice downstream PGT applications. Furthermore, more important than the amplification yield per se are several characteristics of the obtained WGA product: (i) good coverage of the genome, i.e., covering as much as possible of all 3 × 10
9 nucleotides comprising the human genome; (ii) uniformity of the amplified material, i.e., regions of the genome need to be amplified homogeneously to allow the reliable quantification of CNVs; (iii) preserved original nucleotide sequence to enable reliable genotyping, i.e., avoiding loss of one or both gene copies without introducing artificial sequence variation
[11].
In order to amplify the whole genome in a sequence-independent manner, several WGA requirements need to be proposed: (i) use of enzymes with high processivity and high fidelity, i.e., with proofreading activity ensuring lower error rates as well as longer amplicons; (ii) increasing the number of priming events by using universal primers and adjusting annealing/reannealing conditions (i.e., cycling conditions); (iii) reducing the complexity of the genome prior to amplification, e.g., by fragmentation.
3. WGA Types
3.1. PEP-PCR
Zhang et al. first described the application of WGA from a single cell in 1992. They termed this method primer extension preamplification PCR. Their new technique exploited random 15-base oligonucleotides non-specifically binding to a target genome. Theoretically, the primer was composed of a mixture of 1 × 109 different sequences and was estimated to capture and amplify at least 78% of the haploid genome of a single sperm cell as assessed through targeted loci analysis [2]. Initially, the PEP-PCR protocol was claimed to be too lengthy and no better than direct single-cell PCR by other groups [12]. However, following modifications which resulted in a better genome recovery, this protocol has been successfully applied to various cells (amniocytes, chorionic villi, blastomeres) and has enabled several genetic loci of interest in human genetic diseases to be examined with a good amplification efficiency [13][14][15]. The development of this WGA technique was a breakthrough in the field of human PGT, as it allowed for the first time simultaneous multiple locus analysis with the further opportunity to validate the findings. Although contemporary applications do not (or very rarely) employ the original PEP-PCR approach, its principle has been integrated into all successive WGA developments.
3.2. DOP-PCR
Primer extension preamplification was quickly followed by the development of the more widely adopted DOP-PCR protocol, first described by Telenius et al.
[16], to complement the cytogenetic analysis of flow-sorted chromosomes. Degenerate oligonucleotide primer PCR uses partially degenerate primers binding to many sites of the targeted genome. Variations of DOP-PCR primers include oligos with six degenerated bases in the middle, flanked by defined sequences at both ends (traditional DOP-PCR primer 5′-CCGACTCGAGNNNNNNATGTGG-3′) or oligos with a random 3′ end and partially fixed 5′ sequence–oligos with increased degeneracy, also termed tagged random primers. In all cases, due to the primers’ properties, DOP-PCR synthesis occurs in two stages.
3.3. MDA
The next important technical advancement in the application of WGA methods was the development of multiple displacement amplification (MDA). Originally termed multiply-primed rolling circle amplification
[17], MDA was initially developed to amplify circular templates, but was subsequently modified for the amplification of linear ones. MDA exploits the unique properties of bacteriophage φ29 DNA polymerase (phi29)
[18]. This enzyme possesses proofreading activity and has the capacity to perform strand displacement DNA synthesis for more than 70,000 nucleotides under isothermal conditions without dissociating from the template.
3.4. MALBAC
Significantly later, in 2012, Zong et al. reported the development of a new WGA technology termed multiple annealing and looping-based amplification cycles (MALBAC). MALBAC introduces quasi-linear preamplification to reduce the bias associated with non-linear amplification (first stage PCR). The preamplification is initiated with a pool of random primers, each having a common 5′ 27-nucleotide sequence and 3′ eight random nucleotides, hybridizing to the template DNA at low temperatures (15–20 °C, capable of hybridizing at 0 °C)
[19]. Thus, this stage resembles the MDA principle, with the difference that the MALBAC preamplification stage consists of multiple (5 to 12) annealing-extension-denaturation-looping steps, rather than isothermal synthesis
[11].
3.5. Hybrid WGA Methods
As the various WGA technologies were being established, hybrid WGA techniques combining features of PCR-based and MDA-based WGA were also becoming commercially available. The most well-known hybrid WGA methodology is PicoPLEX, originally introduced by Rubicon Genomics and now marketed as SurePlex by Illumina and a few other companies. While information on the exact principle of these commercially available kits is not easily available due to patents and continuous upgrades, they all utilize two-stage PCR. During the first preamplification stage, template DNA is amplified utilizing either: (i) non-self-complementary/self-inert primers which preclude primer dimer formation
[11]; or (ii) primers allowing for the looping of full amplicons, similar to the ones used in MALBAC
[20][21][22].
4. WGA Drawbacks
4.1. ADO and Incomplete Genomic Coverage
It is thought that, once the template DNA concentration falls below a certain input level, the probability of obtaining a complete template genome, especially with the expectation of uniform amplification, decreases dramatically. At very low initial DNA concentrations, random and difficult to predict (stochastic) effects dictate whether a particular genomic region will be amplified or not
[23]. The so-called ‘Monte Carlo effect’ states that “the lower the abundance of any template, the less likely its true abundance will be reflected in the amplified product”
[24]. Incomplete genomic coverage is apparent from such events as allelic or locus dropout—one of the major drawbacks of WGA in human PGT. The phenomenon of ADO is widely recognized—perceived from the first PGT trials performed using direct PCR—and is the main reason for adopting haplotyping—the simultaneous amplification of causative genes together with linked polymorphic markers—as the gold standard in PGT
[5]. It is important to note that ADO can arise not only from incomplete genomic coverage, but also from preferential amplification of one of the alleles. Therefore, ADO is a complex phenomenon not only influenced by the molecular technique, but also inherent to single-cell applications in general.
4.2. Amplification Bias
Amplification bias, also termed PCR drift, results when certain regions/amplicons within a multitemplate reaction are preferentially amplified relative to the entire pool of potential templates
[19]. As expected, the lower the concentration of the initial template, the more prominent the effects of amplification bias. Amplicon representation bias is very much affected by primer composition, i.e., degree of degeneracy of the primer. Substantial overamplification can result due to complementarity between the 3′ region of the primer and the genomic sequence
[19][23].
4.3. Chimera and Non-Template Amplicons
Apart from representation bias, WGA can produce a certain degree of chimera amplicons—a kind of artificial amplicon mapping to different parts of the genome that are not physically linked. The dominant type of chimeras are intra-chromosomal translocations, suggesting that chimeras are produced by neighbouring amplicons randomly connecting on the same chromosome
[25]. While the formation of chimeric PCR fragments has been attributed to MDA
[26] and further demonstrated for other WGA methods
[26][27], no issues arising from this phenomenon have been reported in PGT. Of note, no significant preference has been recorded in the distributions of chimeras and hotspots among chromosomes; however, preferences in overlap length and GC content have been shown to be pertinent to the sequence denaturation temperature, highlighting a direction of action for reducing chimeras
[28]. Non-template amplicons are associated with contamination and are common to any amplification employing random or degenerate oligos
[23]. They can be addressed by implementing good laboratory practices.
4.4. WGA-Independent Improvements
There are a few measures that can be undertaken to increase the efficiency of any WGA technique. The foremost one is to perform a trophectoderm biopsy. Irrespective of the widely demonstrated clinical benefits
[29][30], performing a blastocyst stage biopsy (which implies biopsy of several trophectoderm cells) also affords a number of technical advantages compared to a single-cell biopsy with subsequent WGA. First, in comparison to a single cell, several cells increase the number of template genomes, thus smoothing the abrogating WGA effects described directly above. For example, Handyside and colleagues investigated the relationship between ADO and input cell number. They demonstrated that ADOs occurred randomly at a frequency of approximately 16% in single-cell amplifications, but were undetectable when the number of input cells was increased to 10–20
[31].
5. Performance Comparison of Different WGA Types
5.1. Mappability of Reads
As mentioned earlier, WGA produces a certain fraction of non-specific amplicons. Similarly, using MPS, there will always be a percentage of reads not mapping to the reference genome. The source of these unmappable reads is junk reads arising from the formation of primer dimers, short DNA fragments, and non-target genomes
[27][32]. For example, when comparing the WGA types MDA, MALBAC, and PicoPLEX using 5× sequencing depth, it was demonstrated that the fraction of unmapped reads for all the amplifiers was low (0.035% mean fraction of unmapped reads)
[32].
In contrast, a much higher unmappable read percentage was demonstrated using deeper sequencing (30×). Specifically, the percentage of unmappable reads for GenomePlex (Sigma Aldrich, St. Louis, MO, USA) and PicoPLEX (Rubicon Genomics, Ann Arbor, MI, USA) (which are considered the same chemistry WGA kits) reached 64 and 66%, respectively. REPLI-g MDA kit (Qiagen) and MALBAC (Yikon Genomics, Shanghai, China) showed 18% and 22% of unmapped reads, respectively
[27]. Another group detailed 35–40% of unmappable reads from SurePlex- and MALBAC-amplified single white blood cells (sequencing depth unknown)
[33]. Such large percentages of unmappable reads were proposed to be due to the presence of universal adapter reads, which are WGA-independent, as well as primer sequences and substantial contributions from small amplicons
[27].
5.2. Uniformity of Coverage and CNV Analysis
As discussed earlier, deficient uniformity of coverage is intrinsic to single-cell applications and is further affected by WGA. In PGT-A, it is essential to have uniform coverage or to have bioinformatic algorithms to manage PCR bias.
In 2002, it was reported that DOP-PCR leads to a strong amplification bias, with individual loci differing in copy number by four to six orders of magnitude
[34]. As MDA-based amplification is isothermal, as opposed to PCR-based WGA methods, one common assumption has been that the MDA technique is immune to GC bias. However, DNA regions with a high localized GC content also prove to be problematic for isothermal amplification, leading to reports of under-representation caused by reduced DNA polymerase processivity and poor DNA priming in high GC areas
[23]. Furthermore, it has been shown that the amplification bias in MDA progressively worsens with greater fold amplification, whereas MALBAC and PicoPLEX appear relatively insensitive to reaction gain
[32].
It has subsequently been demonstrated that DOP-PCR and other PCR-based WGA methods exhibit reasonable uniform amplification with reduced regional amplification bias and outperform MDA in terms of CNV detection using arrays or NGS
[32][33].
5.3. Genomic Coverage and SNV Calling
In the era of arrays, genome representation assessment directly depended on array resolution, which, when compared to MPS, could never cover the full genome or exome. Thus, array genome representation percentages cannot be compared to the ones derived from NGS data. Using 10 K SNP arrays, MDA was estimated to amplify 99.82% of the genome
[35]. Using MPS with ~25× mean sequencing depth, MDA covered 72% of the genome and MALBAC achieved up to 93% genomic coverage of single cancer cells
[19]. MDA using either phi29 or
Bst DNA polymerase has been widely reported to achieve a high physical coverage (>90%) from a single-cell genome or exome at a high sequencing depth (typically >30× or at least 15× average sequencing depth)
[25][36][37]. In contrast, GenomePlex and PicoPLEX (kits with the same chemistry) covered only 39% and 52% of the reference genome, respectively (30× sequencing depth)
[27]. Reduced genomic coverage has also been acknowledged for DOP-PCR
[25][36].
Conversely, shallow sequencing depth runs retrieve only very limited fractions of the genome. For example, MDA attained 8.84% genomic coverage at a mean sequencing depth of ~0.5×, which was slightly higher than that of MALBAC (8.06%)
[25].
Taking account of the percentages of genomes retrieved at deep and shallow sequencing depths, it is evident that the WGA methods themselves are capable of amplifying significant proportions of the target genome, and it is in fact the selected parameters of the downstream applications (e.g., MPS) that limit the detection of the amplified genome.
5.4. False Positive Rate
No less important than undetectable alleles are false alleles that occur due to errors made by the DNA polymerase of the WGA or downstream application assay. Usage of high-affinity (not easily dissociated from the DNA strand), robust (working through tough reaction conditions, e.g., without a purification step) and high-fidelity (i.e., maintaining Watson-Crick base pairing) DNA polymerases, in addition to possessing 3′ > 5′ proofreading activity, is the key to reducing the number of false positives during any PCR/DNA synthesis.
6. Comprehensive PGT Solutions Utilizing Different WGA Protocols
6.1. The Beginning of the Massively Parallel Sequencing Era in Human PGT
In the early days of MPS, it became clear that it provides a better signal-to-noise ratio and resolution than aCGH, simply due to advances in technology. NGS specificity for aneuploidy calling was demonstrated to be 99.98% with a sensitivity of 100% [38]. Exceptional multiplexing opportunities and falling NGS costs facilitated the smooth transition of PGT-A towards monumental MPS exploitation. At the beginning of the MPS era in PGT, the majority of applications used SurePlex WGA, as MPS data were often validated by the formerly-used established aCGH protocols that widely employed SurePlex WGA (e.g., [38][39]).
6.2. Karyomapping and Haplarithmisis
Karyomapping was one of the first applications to exploit SNP arrays in PGT. At the time, it was a rapid alternative to the targeted STR typing approach used as standard in PGT-M. Genome-wide SNV haplotyping allows Karyomapping to detect CNVs, meiotic trisomy, monosomy, triploidy, parthenogenetic activation, uniparental heterodisomy, as well as patterns of genomic duplication seen in, for example, hydatidiform moles—all in a single workflow. The assay requires single- or multi-cell embryo biopsy amplified by an isothermal MDA as the starting material
[40][41].
Haplarithmisis—an extension of Karyomapping—is similarly a genome-wide generic PGT tool that originally exploited SNP arrays and MDA for single-/few-cell WGA. A computational pipeline evaluates the observed versus expected SNP probe’s intensity values for each allele in the sample, thus allowing detection of CNVs, the mitotic or meiotic nature of chromosomal anomalies (with the exception of monosomies), low-grade mosaicism, as well as proper ploidy (e.g., enables the distinction of aberrant tetraploid from aberrant diploid)
[42].
6.3. OnePGT
A commercial NGS-based solution that integrates PGT-A, PGT-SR, and PGT-M in a single workflow—OnePGT by Agilent Technologies—has recently been released to the market. The protocol exploits MDA for the embryo WGA and reduced representation WGS and has been verified on a few Illumina-sequencing platforms. To deduce haplotype inheritance, the embedded PGT-M pipeline utilizes principles of Haplarithmisis and was developed by the same group. Both the PGT-A and PGT-SR pipelines are based upon read-count analysis in order to assess CNVs. Inherent to haplotyping methodologies, the processing of additional family members such as the proband or a grandparent is required for haplotype establishment
[43].
6.4. MARSALA
The MPS application termed Mutated Allele Revealed by Sequencing with Aneuploidy and Linkage Analyses (MARSALA) combines low-coverage genome sequencing for PGT-A and the targeted enrichment of mutation loci and linked SNVs for PGT-M. A prerequisite for MARSALA is the genome sequences of the parents. In the absence of an affected relative of the parents, an affected embryo identified by direct calling of the causative SNV or embryo haplotyping can be used as an equivalent of the proband for linkage analyses
[44][45]. The application uses MALBAC for embryo WGA, an aliquot of which is reamplified with a pair of target-specific primers, and then the targeted PCR products are mixed with the native WGA for NGS. In this way, the existence of the point mutation and aneuploidy can be detected in one NGS run. The region of interest can be sequenced to ultra-high coverage (>1000×), still maintaining an accurate CNV measurement throughout the whole genome. It has been demonstrated that, in contrast to MDA, using MALBAC for single-cell WGA linkage analyses can be achieved with only 2× sequencing depth
[44][45].
6.5. MaReCs
Technologically similar to MARSALA and developed by the same group, Mapping Allele with Resolved Carrier Status (MaReCs) is a PGT-SR methodology. Whilst MaReCs does not require pre-clinical work-up to phase haplotypes, it is performed in two stages. First, embryo CNVs are analysed by a high-coverage, high-resolution WGS. Secondly, targeted NGS analysis is performed for 60 adjacent SNVs flanking the translocation breakpoint in a manner similar to MARSALA to perform haplotyping
[46]. The availability of at least one chromosomally imbalanced embryo (so-called reference embryo) is essential for the pipeline to locate a translocation breakpoint. This approach is able to establish whether or not a chromosomally balanced embryo carries the translocation
[46], which is not possible by standard PGT-SR and PGT-A.
6.6. Haploseek
Haploseek is a low-coverage, sequencing-based cPGT application exploiting PicoPLEX for embryo WGA. Although custom target design is not required, a prerequisite for the PGT cycle is pre-case SNP array assessment of the parents and affected child to generate whole genome haplotypes. Further SNP array data, together with sequencing data from the embryos, are integrated using a hidden Markov model, which predicts whether or not the parental-affected haplotypes have been inherited by each of the sequenced embryos across all chromosomes
[47].
6.7. Universal cPGT
Recently, Chen et al. have developed a comprehensive WGS-based PGT tool capable of assessing monogenic disorders, aneuploidies, and chromosomal rearrangements without the requirement of additional family members and without the need of any pre-clinical work-up
[48]. However, PGT-SR can only be performed if unbalanced translocation embryos are available as a reference to distinguish between balanced translocation carriers and normal embryos. Haplotyping for PGT-M is achieved by analysing already-retrieved embryos as a reference, rendering it impossible to analyse cases where direct mutation loci testing in embryos cannot be achieved by NGS (e.g., trinucleotide expansion disorders, intergenic deletions).
7. Conclusion
To enable the genetic diagnosing of preimplantation embryos, all of the current cPGT solutions require clonal amplification of the template DNA. Consequently, WGA is more essential than ever before and has become one of the most important tools in the ever-developing field of human PGT
[23]. The availability of substantial volumes of initially minute amounts of embryonic DNA generated by a single WGA round has made it possible to: (i) avoid embryo rebiopsy and repeat the analysis on account of assay failure or (long term) misdiagnosis or genotype mismatch of the birthed baby; (ii) shorten the turnaround time between referral and clinical cycle because the adaptation/validation of PCR reactions at the single-cell level can be omitted from the pre-clinical work-up
[6]; and, most importantly, (iii) develop multifactor and comprehensive PGT.
MPS-based approaches are much more standardized and allow for high-throughput automation, reduced hands-on time, and minimization of the possibility of human errors—all at a reduced cost
[6]. Hence, MPS-based approaches are regarded as the most powerful platforms for future PGT
[49]. Several groups have already demonstrated the ability to perform mutation loci assessment with a shallow sequencing depth (2–4×) without the need for target enrichment
[44][45][48]. Furthermore, the technical resolution of CNV detection has been demonstrated to be down to several kilobases
[39]. While researchers compete to reduce the testing time and simplify the use of PGT methods, clinically, these objectives are not always justified and can result in painful and hard-to-correct errors. PGT has never been a simple method and, by its very nature, cannot be. Despite all the tempting emerging technical PGT opportunities, clinical PGT should continue to strictly adhere to the existing guidelines
[6][50] and always bear in mind that patient safety is the number one priority. Orthogonal SNV validation should never be omitted unless there is convincing evidence to the contrary. The resolution of CNV detection should not be set unreasonably high to minimize the detection of artifacts resulting from WGA-introduced bias appearing as extensive biological heterogeneity, as this can potentially lead to normal embryos being discarded
[36][39].
 +1 credit
+1 credit