 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tao Yang | -- | 3500 | 2022-07-26 19:20:57 | | | |
| 2 | Peter Tang | Meta information modification | 3500 | 2022-07-27 03:04:44 | | |
Video Upload Options
Acetyl-para-aminophenol (APAP), a commonly used antipyretic analgesic, is becoming increasingly toxic to the liver, resulting in a high rate of acute hepatic failure in Europe and the United States. Excessive APAP metabolism in the liver develops an APAP–protein adduct, which causes oxidative stress, MPTP opening, and hepatic necrosis. HMGB-1, HSP, nDNA, mtDNA, uric acid, and ATP are DMAPs released during hepatic necrosis. DMAPs attach to TLR4-expressing immune cells such KCs, macrophages, and NK cells, activating them and causing them to secrete cytokines. Immune cells and their secreted cytokines have been demonstrated to have a dual function in acetaminophen-induced liver injury (AILI), with a role in either proinflammation or pro-regeneration, resulting in contradicting findings and some research confusion. Neutrophils, KCs, MoMFs, NK/NKT cells, γδT cells, DCs, and inflammasomes have pivotal roles in AILI.
1. Introduction
2. Metabolism of APAP
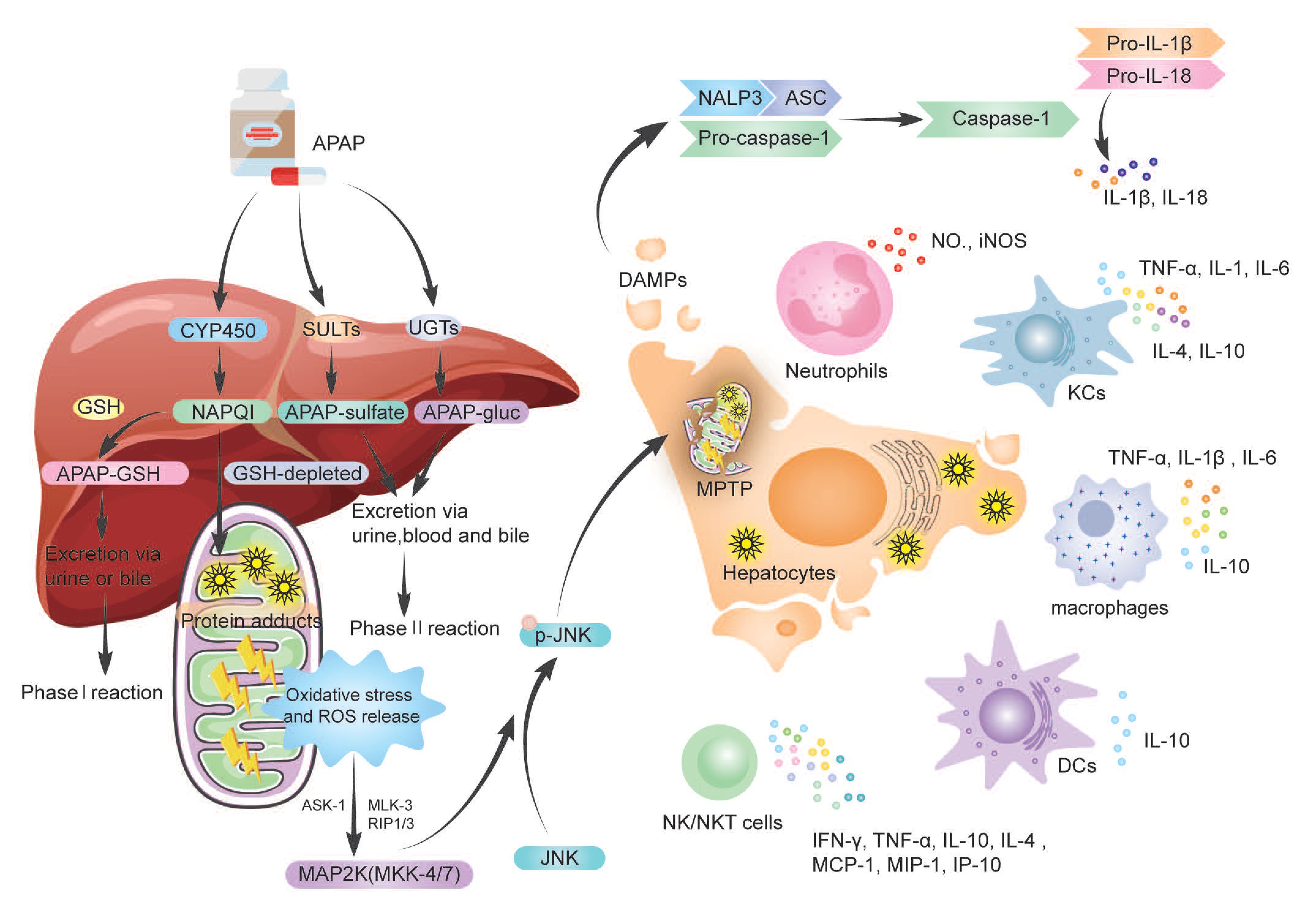
3. The Dual Role of Immune Cells and Cytokine
3.1. Neutrophils in AILI
3.2. Macrophages in AILI
3.3. Dendritic Cells (DCs) in AILI
3.4. Natural Killer Cells (NK Cells) and NKT Cells in AILI
3.5. γδT Cells in AILI
3.6. Cytokine Storm in AILI
References
- Chalasani, N.P.; Maddur, H.; Russo, M.W.; Wong, R.J.; Reddy, K.R. Practice Parameters Committee of the American College of Gas-troenterology. ACG Clinical Guideline: Diagnosis and Management of Idiosyncratic Drug-Induced Liver Injury. Am. J. Gastro-Enterol. 2021, 116, 878–898.
- Real, M.; Barnhill, M.S.; Higley, C.; Rosenberg, J.; Lewis, J.H. Drug-Induced Liver Injury: Highlights of the Recent Literature. Drug Saf. 2019, 42, 365–387.
- Shen, T.; Liu, Y.; Shang, J.; Xie, Q.; Li, J.; Yan, M.; Xu, J.; Niu, J.; Liu, J.; Watkins, P.B.; et al. Incidence and Etiology of Drug-Induced Liver Injury in Mainland China. Gastroenterology. 2019, 156, 2230–2241.
- European Association for the Study of the Liver. Electronic address: ; Clinical Practice Guideline Panel: Chair: Panel members; EASL Governing Board representative: EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 2019, 70, 1222–1261.
- Garcia-Cortes, M.; Robles-Diaz, M.; Stephens, C.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Drug induced liver injury: An up-date. Arch. Toxicol. 2020, 94, 3381–3407.
- Hu, C.; Zhao, L.; Wu, Z.; Li, L. Transplantation of mesenchymal stem cells and their derivatives effectively promotes liver regen-eration to attenuate acetaminophen-induced liver injury. Stem. Cell Res. 2020, 11, 88.
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity-Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331.
- Goldberg, D.S.; Forde, K.A.; Carbonari, D.M.; Lewis, J.D.; Leidl, K.B.; Reddy, K.R.; Haynes, K.; Roy, J.; Sha, D.; Marks, A.R.; et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology 2015, 148, 1353–1361.
- Wei, G.; Bergquist, A.; Broomé, U.; Lindgren, S.; Wallerstedt, S.; Almer, S.; Sangfelt, P.; Danielsson, A.; Sandberg-Gertzén, H.; Lööf, L.; et al. Acute liver failure in Sweden: Etiology and outcome. J. Intern. Med. 2007, 262, 393–401.
- Bernal, W.; Wendon, J. Acute liver failure. N. Engl. J. Med. 2014, 370, 1170–1171.
- Larson, A.M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548.
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405.
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30.
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of aceta-minophen metabolism at the therapeutic versus toxic doses. Pharm. Genom. 2015, 25, 416–426.
- Du, K.; Xie, Y.; McGill, M.R.; Jaeschke, H. Pathophysiological significance of c-jun N-terminal kinase in acetaminophen hepato-toxicity. Expert Opin. Drug Metab. Toxicol 2015, 11, 1769–1779.
- Win, S.; Than, T.A.; Han, D.; Petrovic, L.M.; Kaplowitz, N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acet-aminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J. Biol. Chem. 2011, 286, 35071–35078.
- Win, S.; Than, T.A.; Min, R.W.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial. Src. Hepatol. 2016, 63, 1987–2003.
- Jaeschke, H.; Akakpo, J.Y.; Umbaugh, D.S.; Ramachandran, A. Novel Therapeutic Approaches Against Acetaminophen-induced Liver Injury and Acute Liver Failure. Toxicol. Sci. 2020, 174, 159–167.
- Zhang, J.; Min, R.W.M.; Le, K.; Zhou, S.; Aghajan, M.; Than, T.A.; Win, S.; Kaplowitz, N. The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis. 2017, 8, e2903.
- Zhang, Y.F.; He, W.; Zhang, C.; Liu, X.J.; Lu, Y.; Wang, H.; Zhang, Z.H.; Chen, X.; Xu, D.X. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett. 2014, 225, 445–453.
- Yang, R.; Tonnesseen, T.I. DAMPs and sterile inflammation in drug hepatotoxicity. Hepatol. Int. 2019, 13, 42–50.
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepa-Tol. 2017, 66, 836–848.
- Kono, H.; Chen, C.J.; Ontiveros, F.; Rock, K.L. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Investig. 2010, 120, 1939–1949.
- Amaral, S.S.; Oliveira, A.G.; Marques, P.E.; Pires, D.A.; Resende, R.R.; Sousa, B.R.; Pinto, M.A.; Russo, R.C.; Andrade, L.M.; Leite, M.F.; et al. Altered responsiveness to extracellular ATP enhances acetaminophen hepatotox-icity. Cell Commun. Signal. 2013, 11, 10.
- Guo, H.; Chen, S.; Xie, M.; Zhou, C.; Zheng, M. The complex roles of neutrophils in APAP-induced liver injury. Cell Prolif. 2021, 54, e13040.
- Shen, K.; Chang, W.; Gao, X.; Wang, H.; Niu, W.; Song, L.; Qin, X. Depletion of activated hepatic stellate cell correlates with severe liver damage and abnormal liver regeneration in acetaminophen-induced liver injury. Acta Biochim. Biophys. Sinica 2021, 43, 307–315.
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Im-Munol. 2005, 5, 331–342.
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682.
- Barman, P.K.; Mukherjee, R.; Prusty, B.K.; Suklabaidya, S.; Senapati, S.; Ravindran, B. Chitohexaose protects against acetaminophen-induced hepatotoxicity in mice. Cell Death Dis. 2016, 7, e2224.
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531.
- Jaillon, S.; Galdiero, M.R.; Del Prete, D.; Cassatella, M.A.; Garlanda, C.; Mantovani, A. Neutrophils in innate and adaptive immunity. Semin. Immunopathol. 2013, 35, 377–394.
- Liu, Z.X.; Han, D.; Gunawan, B.; Kaplowitz, N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 2006, 43, 1220–1230.
- Foureau, D.M.; Walling, T.L.; Maddukuri, V.; Anderson, W.; Culbreath, K.; Kleiner, D.E.; Ahrens, W.A.; Jacobs, C.; Watkins, P.B.; Fontana, R.J.; et al. Comparative analysis of portal hepatic infiltrating leucocytes in acute drug-induced liver injury, idiopathic autoimmune and viral hepatitis. Clin. Exp. Immunol. 2015, 180, 40–51.
- Wang, X.; Sun, R.; Wei, H.; Tian, Z. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of γδ T cells with macrophages. Hepatology 2013, 57, 373–384.
- Liu, J.; Jiang, M.; Jin, Q.; Wu, Y.L.; Cui, Z.Y.; Cui, B.W.; Shang, Y.; Zhan, Z.Y.; Lin, Y.C.; Jiao, J.Y.; et al. Modulation of HMGB1 Release in APAP-Induced Liver Injury: A Possible Strategy of Chikusetsusaponin V Targeting NETs Formation. Front. Pharmacol. 2021, 12, 723881.
- Lundbäck, P.; Lea, J.D.; Sowinska, A.; Ottosson, L.; Fürst, C.M.; Steen, J.; Aulin, C.; Clarke, J.I.; Kipar, A.; Klevenvall, L.; et al. A novel high mobility group box 1 neutralizing chimeric antibody attenuates drug-induced liver injury and postinjury inflammation in mice. Hepatology 2016, 64, 1699–1710.
- Ayata, C.K.; Ganal, S.C.; Hockenjos, B.; Willim, K.; Vieira, R.P.; Grimm, M.; Robaye, B.; Boeynaems, J.M.; Di Virgilio, F.; Pellegatti, P.; et al. Purinergic P2Y₂ receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology 2012, 143, 1620–1629.
- He, Y.; Feng, D.; Li, M.; Gao, Y.; Ramirez, T.; Cao, H.; Kim, S.J.; Yang, Y.; Cai, Y.; Ju, C.; et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology 2017, 66, 220–234.
- Volmering, S.; Block, H.; Boras, M.; Lowell, C.A.; Zarbock, A. The Neutrophil Btk Signalosome Regulates Integrin Activation during Sterile Inflammation. Immunity. 2016, 44, 73–87.
- Cover, C.; Liu, J.; Farhood, A.; Malle, E.; Waalkes, M.P.; Bajt, M.L.; Jaeschke, H. Pathophysiological role of the acute inflammatory response during acetaminophen hepa-totoxicity. Toxicol. Appl. Pharm. 2006, 216, 98–107.
- Liu, Z.X.; Govindarajan, S.; Kaplowitz, N. Innate immune system plays a critical role in determining the progression and sever-ity of acetaminophen hepatotoxicity. Gastroenterology 2004, 127, 1760–1774.
- Lawson, J.A.; Farhood, A.; Hopper, R.D.; Bajt, M.L.; Jaeschke, H. The hepatic inflammatory response after acetaminophen over-dose: Role of neutrophils. Toxicol. Sci. 2000, 54, 509–516.
- Williams, C.D.; Bajt, M.L.; Farhood, A.; Jaeschke, H. Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int. 2010, 30, 1280–1292.
- Williams, C.D.; Bajt, M.L.; Sharpe, M.R.; McGill, M.R.; Farhood, A.; Jaeschke, H. Neutrophil activation during acetaminophen hepato-toxicity and repair in mice and humans. Toxicol. Appl. Pharm. 2014, 275, 122–133.
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orches-trate liver repair. Nat. Commun. 2019, 10, 1076.
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Avila, T.V.; Melgaço, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ in-jury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982.
- Graubardt, N.; Vugman, M.; Mouhadeb, O.; Caliari, G.; Pasmanik-Chor, M.; Reuveni, D.; Zigmond, E.; Brazowski, E.; David, E.; Chappell-Maor, L.; et al. Ly6Chi Monocytes and Their Macrophage Descendants Regulate Neutrophil Function and Clearance in Acetaminophen-Induced Liver Injury. Front. Immunol. 2017, 8, 626.
- Jaeschke, H.; Farhood, A.; Smith, C.W. Neutrophil-induced liver cell injury in endotoxin shock is a CD11b/CD18-dependent mechanism. Am. J. Physiol. 1991, 261, G1051–G1056.
- Gong, L.; Liao, L.; Dai, X.; Xue, X.; Peng, C.; Li, Y. The dual role of immune response in acetaminophen hepatotoxicity: Implication for immune pharmacological targets. Toxicol. Lett. 2021, 351, 37–52.
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312.
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743.
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426.
- Jung, H.E.; Kim, T.H.; Lee, H.K. Contribution of Dendritic Cells in Protective Immunity against Respiratory Syncytial Virus In-fection. Viruses 2020, 12, 102.
- Plitas, G.; Burt, B.M.; Stableford, J.A.; Nguyen, H.M.; Welles, A.P.; DeMatteo, R.P. Dendritic cells are required for effective cross-presentation in the murine liver. Hepatology 2008, 47, 1343–1351.
- Pillarisetty, V.G.; Shah, A.B.; Miller, G.; Bleier, J.I.; DeMatteo, R.P. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J. Immunol. 2004, 172, 1009–1017.
- Xia, S.; Guo, Z.; Xu, X.; Yi, H.; Wang, Q.; Cao, X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood 2008, 112, 3175–3185.
- Connolly, M.K.; Bedrosian, A.S.; Malhotra, A.; Henning, J.R.; Ibrahim, J.; Vera, V.; Cieza-Rubio, N.E.; Hassan, B.U.; Pachter, H.L.; Cohen, S.; et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immu-nogenicity. J. Immunol. 2010, 185, 2200–2208.
- Connolly, M.K.; Ayo, D.; Malhotra, A.; Hackman, M.; Bedrosian, A.S.; Ibrahim, J.; Cieza-Rubio, N.E.; Nguyen, A.H.; Henning, J.R.; Dorvil-Castro, M.; et al. Dendritic cell depletion exacerbates acetaminophen hepatotoxicity. Hepatology 2011, 54, 959–968.
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell Mol. Immunol. 2016, 13, 328–336.
- Liu, W.; Zeng, X.; Liu, Y.; Liu, J.; Li, C.; Chen, L.; Chen, H.; Ouyang, D. The Immunological Mechanisms and Immune-Based Biomarkers of Drug-Induced Liver Injury. Front. Pharmacol. 2021, 12, 723940.
- Peng, H.; Jiang, X.; Chen, Y.; Sojka, D.K.; Wei, H.; Gao, X.; Sun, R.; Yokoyama, W.M.; Tian, Z. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J. Clin. Investig. 2013, 123, 1444–1456.
- Wallace, K.L.; Marshall, M.A.; Ramos, S.I.; Lannigan, J.A.; Field, J.J.; Strieter, R.M.; Linden, J. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood 2009, 114, 667–676.
- Fasbender, F.; Obholzer, M.; Metzler, S.; Stöber, R.; Hengstler, J.G.; Watzl, C. Enhanced activation of human NK cells by drug-exposed hepatocytes. Arch. Toxicol. 2020, 94, 439–448.
- Masson, M.J.; Carpenter, L.D.; Graf, M.L.; Pohl, L.R. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology 2008, 48, 889–897.
- Downs, I.; Aw, T.Y.; Liu, J.; Adegboyega, P.; Ajuebor, M.N. Vα14iNKT cell deficiency prevents acetaminophen-induced acute liver failure by enhancing hepatic glutathione and altering APAP metabolism. Biochem. Biophys. Res. Commun. 2012, 428, 245–251.
- Martin-Murphy, B.V.; Kominsky, D.J.; Orlicky, D.J.; Donohue, T.M., Jr.; Ju, C. Increased susceptibility of natural killer T-cell-deficient mice to acetaminophen-induced liver injury. Hepatology 2013, 57, 1575–1584.
- Kwon, H.J.; Won, Y.S.; Park, O.; Feng, D.; Gao, B. Opposing effects of prednisolone treatment on T/NKT cell- and hepatotoxin-mediated hepatitis in mice. Hepatology 2014, 59, 1094–1106.
- Constant, P.; Davodeau, F.; Peyrat, M.A.; Poquet, Y.; Puzo, G.; Bonneville, M.; Fournié, J.J. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science 1994, 264, 267–270.
- Kabelitz, D. Gamma Delta T Cells (γδ T Cells) in Health and Disease: In Memory of Professor Wendy Havran. Cells 2020, 9, 2564.
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100.
- Suzuki, T.; Minagawa, S.; Yamazaki, T.; Arai, T.; Kanai, M.; Shinjo, S.; Goda, N. Loss of hypoxia inducible factor-1α aggravates γδ T-cell-mediated inflammation during acetaminophen-induced liver injury. Hepatol. Commun. 2018, 2, 571–581.
- Ferrara, J.L.; Abhyankar, S.; Gilliland, D.G. Cytokine storm of graft-versus-host disease: A critical effector role for interleukin-1. Transpl. Proc. 1993, 25, 1216–1217.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851.
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273.
- Zhao, J.; Kim, J.W.; Zhou, Z.; Qi, J.; Tian, W.; Lim, C.W.; Han, K.M.; Kim, B. Macrophage-Inducible C-Type Lectin Signaling Exacer-bates Acetaminophen-Induced Liver Injury by Promoting Kupffer Cell Activation in Mice. Mol. Pharmacol. 2021, 99, 92–103.
- Campion, S.N.; Johnson, R.; Aleksunes, L.M.; Goedken, M.J.; van Rooijen, N.; Scheffer, G.L.; Cherrington, N.J.; Manautou, J.E. Hepatic Mrp4 induction following acetaminophen exposure is dependent on Kupffer cell function. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G294–G304.
- Liao, Y.; Yang, Y.; Wang, X.; Wei, M.; Guo, Q.; Zhao, L. Oroxyloside ameliorates acetaminophen-induced hepatotoxicity by inhibiting JNK related apoptosis and necroptosis. J. Ethnopharmacol. 2020, 258, 112917.
- Viswanathan, P.; Sharma, Y.; Jaber, F.L.; Tchaikovskaya, T.; Gupta, S. Transplanted hepatocytes rescue mice in acetaminophen-induced acute liver failure through paracrine signals for hepatic ATM and STAT3 pathways. FASEB J. 2021, 35, e21471.
- Jiang, W.P.; Deng, J.S.; Huang, S.S.; Wu, S.H.; Chen, C.C.; Liao, J.C.; Chen, H.Y.; Lin, H.Y.; Huang, G.J. Sanghuangporus sanghuangMyceli-um Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation. Antioxidants 2021, 10, 897.
- Fisher, J.E.; McKenzie, T.J.; Lillegard, J.B.; Yu, Y.; Juskewitch, J.E.; Nedredal, G.I.; Brunn, G.J.; Yi, E.S.; Malhi, H.; Smyrk, T.C.; et al. Role of Kupffer cells and toll-like receptor 4 in acetaminophen-induced acute liver failure. J. Surg. Res. 2013, 180, 147–155.
- Bourdi, M.; Masubuchi, Y.; Reilly, T.P.; Amouzadeh, H.R.; Martin, J.L.; George, J.W.; Shah, A.G.; Pohl, L.R. Protection against aceta-minophen-induced liver injury and lethality by interleukin 10: Role of inducible nitric oxide synthase. Hepatology 2002, 35, 289–298.
- James, L.P.; Kurten, R.C.; Lamps, L.W.; McCullough, S.; Hinson, J.A. Tumour necrosis factor receptor 1 and hepatocyte regenera-tion in acetaminophen toxicity: A kinetic study of proliferating cell nuclear antigen and cytokine expression. Basic Clin. Pharma-Col. Toxicol. 2005, 97, 8–14.

