Acetyl-para-aminophenol (APAP), a commonly used antipyretic analgesic, is becoming increasingly toxic to the liver, resulting in a high rate of acute hepatic failure in Europe and the United States. Excessive APAP metabolism in the liver develops an APAP–protein adduct, which causes oxidative stress, MPTP opening, and hepatic necrosis. HMGB-1, HSP, nDNA, mtDNA, uric acid, and ATP are DMAPs released during hepatic necrosis. DMAPs attach to TLR4-expressing immune cells such KCs, macrophages, and NK cells, activating them and causing them to secrete cytokines. Immune cells and their secreted cytokines have been demonstrated to have a dual function in acetaminophen-induced liver injury (AILI), with a role in either proinflammation or pro-regeneration, resulting in contradicting findings and some research confusion. Neutrophils, KCs, MoMFs, NK/NKT cells, γδT cells, DCs, and inflammasomes have pivotal roles in AILI.
- innate immune response
- acetyl-para-aminophenol-induced liver injury
- neutrophils
- macrophages
- cytokine
1. Introduction
2. Metabolism of APAP
APAP is a non-steroidal antipyretic and analgesic medication, and the clinically safe dosage varies from 1 to 4 g per day. Following oral treatment, APAP is absorbed from the gut and transported to the liver for metabolism, which requires a variety of enzymatic reactions [11,12][11][12]. The bulk (80–90%) of APAP is metabolized by UDP glucuronosyltransferase (UGTs) and sulfate transferase (SULTs) to inactive glucuronide (APAP-gluc) and sulfate (APAP sulfate), which is then eliminated in the urine, blood and bile, commonly known as the phase II metabolism reaction [13]. The phase I enzymatic reaction occurs when a minor (5–10%) amount of APAP is metabolized in hepatocytes by cytochrome P450 (CYP450) enzymes, such as CYP2E1 and CYE1A2, to the reactive metabolite N-acetyl-p-benzoquinone imine (NAPQI). The hepatic antioxidant glutathione (GSH) rapidly converts NAPQI to a harmless form via glutathione-S-transferase in an enzymatic process, forming the APAP-GSH complex (APAP-GSH), which is further metabolized to N-acetyl-L-cysteine adducts, cysteamide adducts, and cysteamide/glycine adducts, excreted through the urine or bile [14]. When the phase II reaction is saturated, excessive APAP is metabolized by the phase I reaction, NAPQI accumulation (Figure 1). When GSH is depleted, the growing concentration of NAPQI forms harmful APAP protein adducts, also known as NAPQI–protein adduct peptides, by covalently reacting with protein sulfur groups. In conclusion, both the generation of APAP protein adducts and the depletion of GSH result in increased protein adduct formation in hepatocytes, resulting in oxidative stress and increased reactive oxygen species (ROS) production. However, in the mitochondria of hepatocytes, those protein adducts appear to trigger limited oxidative stress, which is responsible for the induction of mitogen-activated protein kinase (MAPK); then, MAPK kinases (MKK)-4/7 activate c-Jun N-terminal kinase (JNK), leading to phosphorylation of JNK in the cytosol [15]. Then, p-JNK is translocated to mitochondria, where it binds the outer mitochondrial membrane anchor protein Sab (SH3-domain-binding protein that preferentially associates with Btk) or Sh3bp5 [16], generating dephosphorylation of intermembrane Src [17]. Thus, activated JNK and mitochondria p-JNK translocation, in turn, further enhances oxidative stress signal to induce the mitochondrial membrane permeability transition pore (MPTP) opening, causing mitochondrial membrane permeability and dysfunction [18], ultimately leading to hepatocyte necrosis and liver failure. This process needs various kinases involved, for example, mitochondria activating MAP3 kinases (MAP3K) such as maxed-lineage kinase-3 (MLK-3), apoptosis signal-regulating kinase-1 (ASK-1), and MAP2K [19], receptor-interacting protein 1(RIP1), and RIP3 [20]. However, more research is needed to magnify the understanding of the comprehensive relationship between kinases and JNK signals.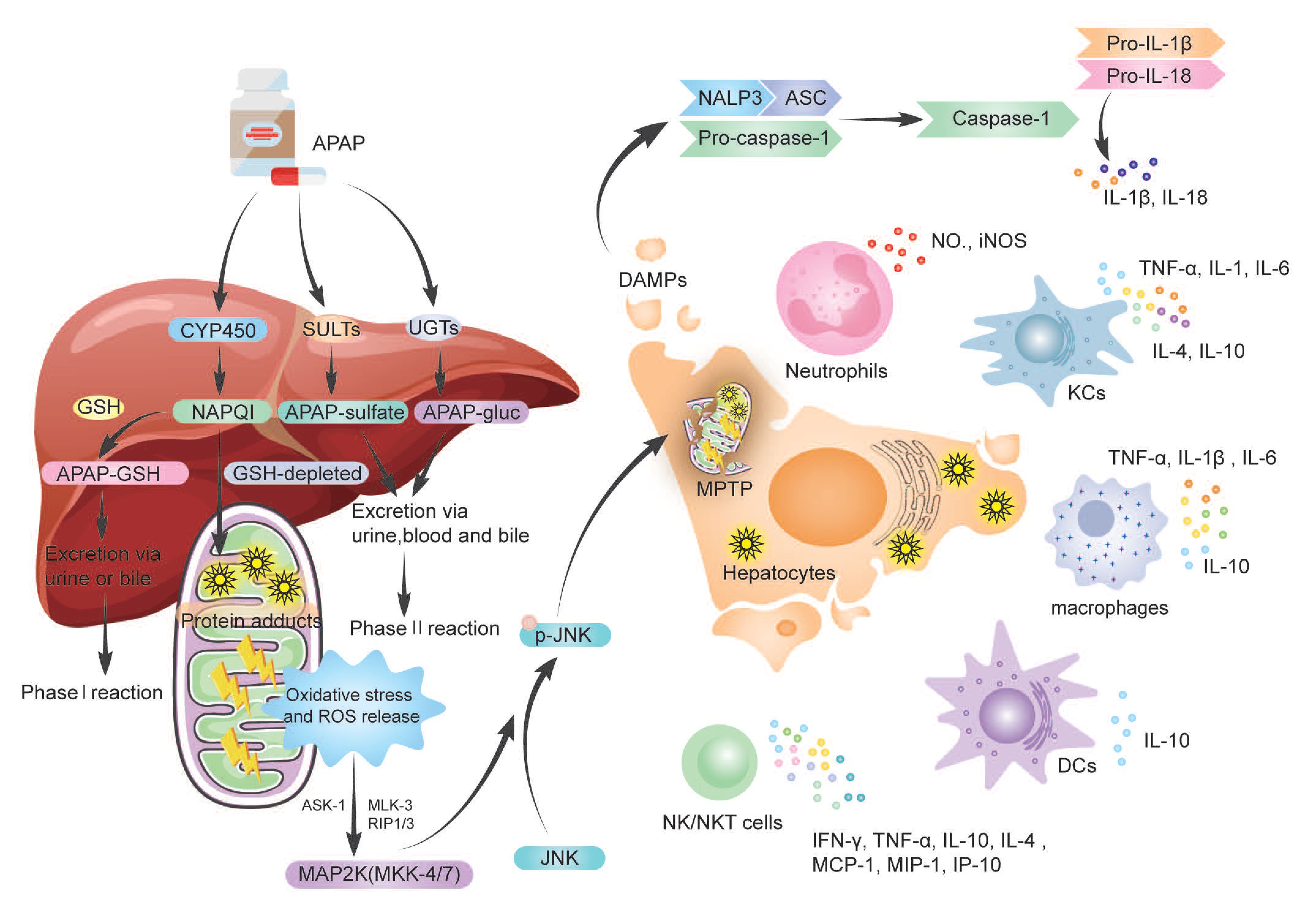
3. The Dual Role of Immune Cells and Cytokine
3.1. Neutrophils in AILI
Neutrophils are the first-line defense of the innate immune response, which is the mighty capacity for biosynthetic activity, including complement components, Fc receptors, cytokines, and chemokines [30]. Neutrophils usually are inactivated and travel slowly and aimlessly through the peripheral blood circulation. When the pathogen invades or endogenous stimulants are released, pattern recognition receptors can identify pathogen-associated molecular patterns (PAMPs) and DAMPs. The neutrophils in circulation are activated, which migrate to the injury site [31]. The study revealed that the recruited neutrophils (Mac-1+ Gr-1+) significantly increased in the liver at 6 and 24 h after APAP treatment, and during the recovery period, neutrophils subsequently declined [32]. How do neutrophils recruit to the area of hepatocyte necrosis? First is the release of various endogenous DAMPs by necrotic hepatocytes, as demonstrated in liver biopsies [33]. The HMGB1-TLR4-IL-23-IL-17A axis can facilitate neutrophil penetration after APAP treatment, whereas the HMGB1 inhibitor glycyrrhizin drastically inhibits IL-23 and IL-17A production as well as hepatic neutrophil accumulation [34]. Further study by L et al. found that APAP increases HMGB1 expression via activating Caspase-1 in hepatocytes, while neutrophil depletion or abolishing neutrophil extracellular traps (NETs) formation reduces HMGB1 levels and prevents hepatocyte necrosis [35]. Therefore, those studies confirmed that HMGB1 is an indeed targeted DAMP. The partly humanized anti-HMGB1 monoclonal antibody (mAb; h2G7), as compared with NAC, has high therapeutic efficacy and an extended therapeutic window in APAP-ALI [36]. Other DAMPs, such as the release of endogenous ATP, activate the purinergic P2 receptor (P2R) [37] and uric acid [23], promoting neutrophil infiltration and subsequent mouse hepatocyte death; mtDNA activates neutrophils by binding to TLR-9 [38]. In addition to DAMPs, other molecules, such as CXC chemokine receptor 2 (CXCR2), formylated peptide [39], and intercellular adhesion molecule-1 (ICAM-1) [40] could rapidly attract neutrophils to sites of hepatocytes necrosis. The third is other immune cells, such as NK cells and NKT cells, regulating neutrophil accumulation by secreting INF-γ [41]. However, whether the recruited neutrophils have an injurious or protective role in AILI is controversial. It has been hypothesized that neutrophil infiltration protects against hepatic necrosis and promotes liver healing. Hepatic damage has been associated with neutrophil recruitment in studies. There was no increase in Mac-1 (CD11b/CD18) expression or L-selectin shedding on circulating neutrophils, and anti-CD18 antibodies were not protective against AILI in the first 24 h [42]. This neutrophil infiltration was dominated by removing necrotic cell debris but was not robust enough to cause further damage. Further studies found that CD18-deficient mice did not differ from wild-type mice regarding an inflammatory response or liver injury in AILI [43]. Neutrophil depletion by anti-neutrophil antibody Gr-1, gp91phox−/− (an essential subunit of NADPH oxidase, a significant source of phagocytic superoxide) [44], anti-Ly6G antibody, genetic knockout in granulocyte colony-stimulating factor, or genetic deletion in NADPH oxidase 2 (Nox2) [45] did not protect against APAP hepatotoxicity. Those studies suggested that AILI is dominated by intracellular cell death mechanisms in mice rather than being neutrophil-mediated [43]. The possible reason is that neutrophils are not activated in the early stages of AILI and fail to induce injury. Instead, the activation status (CD11b expression and ROS priming) during and after the damage peak [44]. Then, the neutrophils promote the phenotypic conversion of proinflammatory Ly6ChiCX3CR1lo monocytes/macrophages to pro-resolving Ly6CloCX3CR1hi macrophages by expressing reactive oxygen species (ROS), promoting liver repair after APAP injury [45]. In APAP overdose, it has also been suggested that neutrophils cause liver impairment. Liu et al. used an anti-Gr-1 antibody (RB6-8C5) to deplete neutrophils in vivo, which effectively protected against AILI in mice model through reduced FasL expression, direct hepatocytotoxicity, and mitochondrial respiratory chain burst (NO, iNOS) in hepatic leukocytes [32], moreover, ICAM-1 was also involved in the injury process, and AILI was significantly reduced in ICAM-1 deficient mouse. Another study blockade of neutrophil infiltration by anti-granulocyte receptor 1 depletion or combined CXCR2-FPR1 antagonism significantly prevented liver injury after APAP overdose [46]. Both studies used anti-neutrophil antibodies, which selectively deplete circulating neutrophils, causing most neutrophils to wedge in the capillary bed, where they were recognized and phagocytosed by KCs in the liver activated KCs. Activated KCs amplify the inflammatory response and exacerbate the injury by releasing cytokines through phagocytosis. The precise mechanisms of action of neutrophils in AILI are challenging since the exact process by which circulating neutrophils are entirely depleted and interact with other immune cells is unknown. During the injury and recovery stages of AILI, the infiltrating Ly6Chi monocytes, their macrophage descendants, and neutrophils spatially and temporally overlap in the centrilobular necrotic areas [47]. A decrease in neutrophils that produce ROS has been observed following inducible ablation of circulating Ly6Chi monocytes [47]. On the other hand, these controversial opinions may be associated with the experimental protocol of the researcher. Anti-CD18 antibodies, another example, were previously shown to reduce neutrophils by only 50% in an endotoxin shock model [48], which could explain why mice treated with anti-CD18 antibodies are not protected against AILI; similarly, CD18-deficient, neutrophil-specific antibody Ly6G or congenital neutropenia did not affect the AILI. Because partial or selective inhibition may lead to debatable findings, complete eradication of neutrophils and their function is required to account for the crucial role of neutrophils in AILI [49]. Lastly, the experimental observation time point may be relevant. AILI is a progressive process; for example, mechanisms of injury caused by different immune cells recruited at other time points may diverge, so investigators may need to study injury mechanisms at several different time points within 24 h and continually evaluate them.3.2. Macrophages in AILI
Hepatic macrophages belong to the mononuclear macrophage system and respond to various liver injury signals. The macrophages observed in the currently damaged liver are heterogeneous and have two primary sources: liver-resident macrophages, named Kupffer cells (KCs), which are long-lived, self-renewing, and non-migratory macrophages [74][50]; the other source is blood/bone marrow monocyte-derived macrophages (MoMFs) and peritoneum macrophages, which identify danger signals, such as cytokines and chemokines, migrating to the liver. Liver macrophages that originate from various sources differ in activation and function, with a remarkably flexible and phenotypic alteration. Their functions are multidimensional and even opposing, directly influencing the outcome of the immune response [75][51].3.3. Dendritic Cells (DCs) in AILI
Dendritic cells (DCs), as the most important antigen-presenting cells (APC), are a critical link between innate and adaptive immunity [93][52]. According to the different differentiation pathways, human DCs originated from hematopoietic stem cells, and divided into myeloid DCs (mDCs) and plasmacytoid DCs(pDCs) [94][53]. Even though most DCs are immature in hepatic, a small part of CD11chi conventional DCs (cDCs) express high levels of costimulatory molecules [95][54]; therefore, DCs are more commonly mediate tolerance rather than immunogenicity in the liver [96,97][55][56]. When the liver is wounded, DCs shift their immunological phenotype and become highly immunogenic, modulating NK cells and activating T cells through the production of TNF-α [98][57]. In AILI, DCs reveal a protective role. Connolly et al. found that liver DCs immune-phenotype was markedly altered after APAP treatment, which expressed higher MHC II, costimulatory molecules, and TLR, and produced IL-6, monocyte chemoattractant protein-1 (MCP-1), and TNF-α [68][58]. In the AILI model, depleted DCs aggravated liver necrosis and increased mouse mortality; on the contrary, endogenous DCs expansion using FMS-like tyrosine kinase 3 ligand (Flt3L) blocked AILI progression in mice. The protective mechanism may prevent NKs cell activation and induce neutrophil apoptosis [68][58]. Due to the role of DCs in AILI being less studied, the dual of DCS needs to be further investigated.3.4. Natural Killer Cells (NK Cells) and NKT Cells in AILI
Innate lymphocytes, such as NK cells, NKT cells, and γδT cells, as well as adaptive lymphocytes, such as αβT cells and B cells, are stored in the liver. NK cells, NKT cells, and T cells are up to 65% of all hepatic lymphocytes in humans [99][59], with NK cells accounting for 30–50% [100][60]. The liver NK cells can divide into two subsets, CD49a−DX5+ (conventional NK cells, cNK cells) and CD49a+DX5− (the liver-resident NK cells) [101][61]. NKT cells are a subpopulation of T cells that express both NK cell receptors and T cell receptors, and are MHC I–like molecules, are CD1d-restricted, and are glycolipid antigen reactive [102][62]. Activated NKs and NKTs produce inflammatory mediators such as interferon (IFN)-γ, TNF-α, IL-10, and IL-4, then induce apoptosis. Activated NK cell ligand-protein levels were significantly increased after drug exposure and activated NK cells exacerbated hepatocytotoxicity by secreting IFN-α. In contrast, specific antibodies for NK cell receptors attenuated drug hepatotoxicity [103][63]. It has been reported that activation of NKs and NKTs amplifies the immune response and leads to the exacerbation of AILI by upregulating IFN-γ, FasL-Keratinocyte-derived chemokine (KC), IP-10 (interferon-inducible protein), Mig (monokine induced by IFN-gamma), MCP-1, macrophage inflammatory protein (MIP)-1α, and increased neutrophil accumulation in the liver [41]. Indeed, the pathogenic effects of NKT cells and NK cells in AILI in mice were dependent on the presence of Dimethyl sulfoxide (DMSO), and DMSO-activated hepatic NKT cells and NK cells in vivo, as evidenced by increased numbers of NKT cells and increased intracellular levels of the cytotoxic effector molecules IFN-γ and granzyme B [69][64]. Depletion of NK cells and NKT cells with NK1.1 antibodies attenuate AILI [41]. Similarly, Vα14iNKT-cells-deficient (Jα18−/−) mice could be resistant to AILI, possibly related to APAP metabolic alterations resulting in reduced hepatic GSH binding and glucuronide binding [70][65]. However, studies have also shown that NK/NKT cells have a protective effect against AILI. For example, NKT-cells-deficient mice (CD1d−/− and Jα18−/−) were more vulnerable to AILI than wild-type animals, by a mechanism that involves upregulation and activation of CYP2E1 expression, ultimately leading to APAP protein adduct formation [71][66]. NKT cells also mediate endogenous IL-4 production, while glutathione synthesis is regulated by endogenous IL-4 under stress conditions to control the severity of AILI [71][66]. Kwon et al. also found that NKT cells are beneficial in AILI, but they also reduce the release of inflammatory cytokines [72][67]. Currently, there is no definitive research on the effect of NK/NKT cells in AILI.3.5. γδT Cells in AILI
γδT cells are another type of lymphocyte that plays an essential role in immune response and immunopathological processes and have received widespread attention [104,105][68][69]. Like NKT cells, γδT cells are innate-like T cells, which are an integral component of innate immunity and play an essential role in killing infected or damaged cells and regulating functions of innate cells [106][70]. It was found that IL-17A+ CD3+ γδT cell receptor (TCR) (+) cells were significantly increased in the AILI model, and depletion of γδT cells significantly reduced IL-17A production and attenuated liver injury by reducing hepatic neutrophil recruitment; further studies revealed that γδT cells were activated by macrophage-derived IL-1β and IL-23 [34]. Hypoxia-inducible factor-1 (HIF) expression in T cells attenuated abnormal γδT cell recruitment and alleviated APAP-induced acute inflammatory response, thereby reducing neutrophil infiltration in the liver [73][71]. Both studies suggest that γδT cells play a traumatic role in AILI; however, the specific mechanism of injury remains to be further studied.3.6. Cytokine Storm in AILI
Cytokine storm was first described in 1993 [107][72] that developed after chimeric antigen receptor (CAR) T-cell therapy [108][73]. David C. illustrates that cytokine storm and cytokine release syndrome (CRS) were life-threatening systemic inflammatory syndromes involving elevated levels of circulating cytokines and immunity caused by various therapies, pathogens, cancer, autoimmune diseases, and single-gene diseases [109][74]. There is no doubt that the changes in serum cytokines in AILI patients and animal models may contribute to the development of systemic inflammation and may even lead to acute liver failure or MOF. In AILI, necrotic hepatocytes release DMAPs to recruit immune cells, subsequently activating immune cells to release cytokines (TNF-α, IL-1β, IL-6) and chemokines (MCP-1). The released cytokines then are involved in inflammatory responses or immune-mediated liver injury, resulting in a vicious cycle between cytokines and liver damage. Why do cytokines appear to have opposite results in AILI? First, the mechanism of cytokine secretion is complex, and the same cytokine may be secreted by several immune cells. For example, TNF-α can be secreted by KCs, macrophages, and DC cells; the mechanism of KCs in AILI is oppositional [52,54][75][76]. Second, cytokine secretion is also regulated by cellular signaling pathways, such as JNK [116][77], STAT3 [117][78], MAPK [118][79], and TLR4 [50][80] signaling pathways. Lastly, there may be inconsistencies between animal models and patients with APAP overdose. For instance, in animal experiments, IL-10-deficiency-enhanced cytokine secretion (e.g., TNF-α, IL-1α) and iNOS expression have been observed [121][81]. At the same time, a minor increase in plasma IL-10 has been observed in patients with APAP overdose, and there was no relationship between IL-10 concentrations and the severity of the hepatic injury [122][82]. Therefore, the mechanisms of cytokine secretion, regulation, and effects are complex and need to be further investigated.References
- Chalasani, N.P.; Maddur, H.; Russo, M.W.; Wong, R.J.; Reddy, K.R. Practice Parameters Committee of the American College of Gas-troenterology. ACG Clinical Guideline: Diagnosis and Management of Idiosyncratic Drug-Induced Liver Injury. Am. J. Gastro-Enterol. 2021, 116, 878–898.
- Real, M.; Barnhill, M.S.; Higley, C.; Rosenberg, J.; Lewis, J.H. Drug-Induced Liver Injury: Highlights of the Recent Literature. Drug Saf. 2019, 42, 365–387.
- Shen, T.; Liu, Y.; Shang, J.; Xie, Q.; Li, J.; Yan, M.; Xu, J.; Niu, J.; Liu, J.; Watkins, P.B.; et al. Incidence and Etiology of Drug-Induced Liver Injury in Mainland China. Gastroenterology. 2019, 156, 2230–2241.
- European Association for the Study of the Liver. Electronic address: ; Clinical Practice Guideline Panel: Chair: Panel members; EASL Governing Board representative: EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 2019, 70, 1222–1261.
- Garcia-Cortes, M.; Robles-Diaz, M.; Stephens, C.; Ortega-Alonso, A.; Lucena, M.I.; Andrade, R.J. Drug induced liver injury: An up-date. Arch. Toxicol. 2020, 94, 3381–3407.
- Hu, C.; Zhao, L.; Wu, Z.; Li, L. Transplantation of mesenchymal stem cells and their derivatives effectively promotes liver regen-eration to attenuate acetaminophen-induced liver injury. Stem. Cell Res. 2020, 11, 88.
- Lee, W.M. Acetaminophen (APAP) hepatotoxicity-Isn’t it time for APAP to go away? J. Hepatol. 2017, 67, 1324–1331.
- Goldberg, D.S.; Forde, K.A.; Carbonari, D.M.; Lewis, J.D.; Leidl, K.B.; Reddy, K.R.; Haynes, K.; Roy, J.; Sha, D.; Marks, A.R.; et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology 2015, 148, 1353–1361.
- Wei, G.; Bergquist, A.; Broomé, U.; Lindgren, S.; Wallerstedt, S.; Almer, S.; Sangfelt, P.; Danielsson, A.; Sandberg-Gertzén, H.; Lööf, L.; et al. Acute liver failure in Sweden: Etiology and outcome. J. Intern. Med. 2007, 262, 393–401.
- Bernal, W.; Wendon, J. Acute liver failure. N. Engl. J. Med. 2014, 370, 1170–1171.
- Larson, A.M. Acetaminophen hepatotoxicity. Clin. Liver Dis. 2007, 11, 525–548.
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of acetaminophen-induced liver necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405.
- Ramachandran, A.; Jaeschke, H. Acetaminophen Toxicity: Novel Insights into Mechanisms and Future Perspectives. Gene Expr. 2018, 18, 19–30.
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of aceta-minophen metabolism at the therapeutic versus toxic doses. Pharm. Genom. 2015, 25, 416–426.
- Du, K.; Xie, Y.; McGill, M.R.; Jaeschke, H. Pathophysiological significance of c-jun N-terminal kinase in acetaminophen hepato-toxicity. Expert Opin. Drug Metab. Toxicol 2015, 11, 1769–1779.
- Win, S.; Than, T.A.; Han, D.; Petrovic, L.M.; Kaplowitz, N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acet-aminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J. Biol. Chem. 2011, 286, 35071–35078.
- Win, S.; Than, T.A.; Min, R.W.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial. Src. Hepatol. 2016, 63, 1987–2003.
- Jaeschke, H.; Akakpo, J.Y.; Umbaugh, D.S.; Ramachandran, A. Novel Therapeutic Approaches Against Acetaminophen-induced Liver Injury and Acute Liver Failure. Toxicol. Sci. 2020, 174, 159–167.
- Zhang, J.; Min, R.W.M.; Le, K.; Zhou, S.; Aghajan, M.; Than, T.A.; Win, S.; Kaplowitz, N. The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis. 2017, 8, e2903.
- Zhang, Y.F.; He, W.; Zhang, C.; Liu, X.J.; Lu, Y.; Wang, H.; Zhang, Z.H.; Chen, X.; Xu, D.X. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett. 2014, 225, 445–453.
- Yang, R.; Tonnesseen, T.I. DAMPs and sterile inflammation in drug hepatotoxicity. Hepatol. Int. 2019, 13, 42–50.
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepa-Tol. 2017, 66, 836–848.
- Kono, H.; Chen, C.J.; Ontiveros, F.; Rock, K.L. Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Investig. 2010, 120, 1939–1949.
- Amaral, S.S.; Oliveira, A.G.; Marques, P.E.; Pires, D.A.; Resende, R.R.; Sousa, B.R.; Pinto, M.A.; Russo, R.C.; Andrade, L.M.; Leite, M.F.; et al. Altered responsiveness to extracellular ATP enhances acetaminophen hepatotox-icity. Cell Commun. Signal. 2013, 11, 10.
- Guo, H.; Chen, S.; Xie, M.; Zhou, C.; Zheng, M. The complex roles of neutrophils in APAP-induced liver injury. Cell Prolif. 2021, 54, e13040.
- Shen, K.; Chang, W.; Gao, X.; Wang, H.; Niu, W.; Song, L.; Qin, X. Depletion of activated hepatic stellate cell correlates with severe liver damage and abnormal liver regeneration in acetaminophen-induced liver injury. Acta Biochim. Biophys. Sinica 2021, 43, 307–315.
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Im-Munol. 2005, 5, 331–342.
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682.
- Barman, P.K.; Mukherjee, R.; Prusty, B.K.; Suklabaidya, S.; Senapati, S.; Ravindran, B. Chitohexaose protects against acetaminophen-induced hepatotoxicity in mice. Cell Death Dis. 2016, 7, e2224.
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531.
- Jaillon, S.; Galdiero, M.R.; Del Prete, D.; Cassatella, M.A.; Garlanda, C.; Mantovani, A. Neutrophils in innate and adaptive immunity. Semin. Immunopathol. 2013, 35, 377–394.
- Liu, Z.X.; Han, D.; Gunawan, B.; Kaplowitz, N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 2006, 43, 1220–1230.
- Foureau, D.M.; Walling, T.L.; Maddukuri, V.; Anderson, W.; Culbreath, K.; Kleiner, D.E.; Ahrens, W.A.; Jacobs, C.; Watkins, P.B.; Fontana, R.J.; et al. Comparative analysis of portal hepatic infiltrating leucocytes in acute drug-induced liver injury, idiopathic autoimmune and viral hepatitis. Clin. Exp. Immunol. 2015, 180, 40–51.
- Wang, X.; Sun, R.; Wei, H.; Tian, Z. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of γδ T cells with macrophages. Hepatology 2013, 57, 373–384.
- Liu, J.; Jiang, M.; Jin, Q.; Wu, Y.L.; Cui, Z.Y.; Cui, B.W.; Shang, Y.; Zhan, Z.Y.; Lin, Y.C.; Jiao, J.Y.; et al. Modulation of HMGB1 Release in APAP-Induced Liver Injury: A Possible Strategy of Chikusetsusaponin V Targeting NETs Formation. Front. Pharmacol. 2021, 12, 723881.
- Lundbäck, P.; Lea, J.D.; Sowinska, A.; Ottosson, L.; Fürst, C.M.; Steen, J.; Aulin, C.; Clarke, J.I.; Kipar, A.; Klevenvall, L.; et al. A novel high mobility group box 1 neutralizing chimeric antibody attenuates drug-induced liver injury and postinjury inflammation in mice. Hepatology 2016, 64, 1699–1710.
- Ayata, C.K.; Ganal, S.C.; Hockenjos, B.; Willim, K.; Vieira, R.P.; Grimm, M.; Robaye, B.; Boeynaems, J.M.; Di Virgilio, F.; Pellegatti, P.; et al. Purinergic P2Y₂ receptors promote neutrophil infiltration and hepatocyte death in mice with acute liver injury. Gastroenterology 2012, 143, 1620–1629.
- He, Y.; Feng, D.; Li, M.; Gao, Y.; Ramirez, T.; Cao, H.; Kim, S.J.; Yang, Y.; Cai, Y.; Ju, C.; et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology 2017, 66, 220–234.
- Volmering, S.; Block, H.; Boras, M.; Lowell, C.A.; Zarbock, A. The Neutrophil Btk Signalosome Regulates Integrin Activation during Sterile Inflammation. Immunity. 2016, 44, 73–87.
- Cover, C.; Liu, J.; Farhood, A.; Malle, E.; Waalkes, M.P.; Bajt, M.L.; Jaeschke, H. Pathophysiological role of the acute inflammatory response during acetaminophen hepa-totoxicity. Toxicol. Appl. Pharm. 2006, 216, 98–107.
- Liu, Z.X.; Govindarajan, S.; Kaplowitz, N. Innate immune system plays a critical role in determining the progression and sever-ity of acetaminophen hepatotoxicity. Gastroenterology 2004, 127, 1760–1774.
- Lawson, J.A.; Farhood, A.; Hopper, R.D.; Bajt, M.L.; Jaeschke, H. The hepatic inflammatory response after acetaminophen over-dose: Role of neutrophils. Toxicol. Sci. 2000, 54, 509–516.
- Williams, C.D.; Bajt, M.L.; Farhood, A.; Jaeschke, H. Acetaminophen-induced hepatic neutrophil accumulation and inflammatory liver injury in CD18-deficient mice. Liver Int. 2010, 30, 1280–1292.
- Williams, C.D.; Bajt, M.L.; Sharpe, M.R.; McGill, M.R.; Farhood, A.; Jaeschke, H. Neutrophil activation during acetaminophen hepato-toxicity and repair in mice and humans. Toxicol. Appl. Pharm. 2014, 275, 122–133.
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orches-trate liver repair. Nat. Commun. 2019, 10, 1076.
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Avila, T.V.; Melgaço, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ in-jury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982.
- Graubardt, N.; Vugman, M.; Mouhadeb, O.; Caliari, G.; Pasmanik-Chor, M.; Reuveni, D.; Zigmond, E.; Brazowski, E.; David, E.; Chappell-Maor, L.; et al. Ly6Chi Monocytes and Their Macrophage Descendants Regulate Neutrophil Function and Clearance in Acetaminophen-Induced Liver Injury. Front. Immunol. 2017, 8, 626.
- Jaeschke, H.; Farhood, A.; Smith, C.W. Neutrophil-induced liver cell injury in endotoxin shock is a CD11b/CD18-dependent mechanism. Am. J. Physiol. 1991, 261, G1051–G1056.
- Gong, L.; Liao, L.; Dai, X.; Xue, X.; Peng, C.; Li, Y. The dual role of immune response in acetaminophen hepatotoxicity: Implication for immune pharmacological targets. Toxicol. Lett. 2021, 351, 37–52.
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312.
- Guillot, A.; Tacke, F. Liver Macrophages: Old Dogmas and New Insights. Hepatol. Commun. 2019, 3, 730–743.
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426.
- Jung, H.E.; Kim, T.H.; Lee, H.K. Contribution of Dendritic Cells in Protective Immunity against Respiratory Syncytial Virus In-fection. Viruses 2020, 12, 102.
- Plitas, G.; Burt, B.M.; Stableford, J.A.; Nguyen, H.M.; Welles, A.P.; DeMatteo, R.P. Dendritic cells are required for effective cross-presentation in the murine liver. Hepatology 2008, 47, 1343–1351.
- Pillarisetty, V.G.; Shah, A.B.; Miller, G.; Bleier, J.I.; DeMatteo, R.P. Liver dendritic cells are less immunogenic than spleen dendritic cells because of differences in subtype composition. J. Immunol. 2004, 172, 1009–1017.
- Xia, S.; Guo, Z.; Xu, X.; Yi, H.; Wang, Q.; Cao, X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood 2008, 112, 3175–3185.
- Connolly, M.K.; Bedrosian, A.S.; Malhotra, A.; Henning, J.R.; Ibrahim, J.; Vera, V.; Cieza-Rubio, N.E.; Hassan, B.U.; Pachter, H.L.; Cohen, S.; et al. In hepatic fibrosis, liver sinusoidal endothelial cells acquire enhanced immu-nogenicity. J. Immunol. 2010, 185, 2200–2208.
- Connolly, M.K.; Ayo, D.; Malhotra, A.; Hackman, M.; Bedrosian, A.S.; Ibrahim, J.; Cieza-Rubio, N.E.; Nguyen, A.H.; Henning, J.R.; Dorvil-Castro, M.; et al. Dendritic cell depletion exacerbates acetaminophen hepatotoxicity. Hepatology 2011, 54, 959–968.
- Peng, H.; Wisse, E.; Tian, Z. Liver natural killer cells: Subsets and roles in liver immunity. Cell Mol. Immunol. 2016, 13, 328–336.
- Liu, W.; Zeng, X.; Liu, Y.; Liu, J.; Li, C.; Chen, L.; Chen, H.; Ouyang, D. The Immunological Mechanisms and Immune-Based Biomarkers of Drug-Induced Liver Injury. Front. Pharmacol. 2021, 12, 723940.
- Peng, H.; Jiang, X.; Chen, Y.; Sojka, D.K.; Wei, H.; Gao, X.; Sun, R.; Yokoyama, W.M.; Tian, Z. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J. Clin. Investig. 2013, 123, 1444–1456.
- Wallace, K.L.; Marshall, M.A.; Ramos, S.I.; Lannigan, J.A.; Field, J.J.; Strieter, R.M.; Linden, J. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood 2009, 114, 667–676.
- Fasbender, F.; Obholzer, M.; Metzler, S.; Stöber, R.; Hengstler, J.G.; Watzl, C. Enhanced activation of human NK cells by drug-exposed hepatocytes. Arch. Toxicol. 2020, 94, 439–448.
- Masson, M.J.; Carpenter, L.D.; Graf, M.L.; Pohl, L.R. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology 2008, 48, 889–897.
- Downs, I.; Aw, T.Y.; Liu, J.; Adegboyega, P.; Ajuebor, M.N. Vα14iNKT cell deficiency prevents acetaminophen-induced acute liver failure by enhancing hepatic glutathione and altering APAP metabolism. Biochem. Biophys. Res. Commun. 2012, 428, 245–251.
- Martin-Murphy, B.V.; Kominsky, D.J.; Orlicky, D.J.; Donohue, T.M., Jr.; Ju, C. Increased susceptibility of natural killer T-cell-deficient mice to acetaminophen-induced liver injury. Hepatology 2013, 57, 1575–1584.
- Kwon, H.J.; Won, Y.S.; Park, O.; Feng, D.; Gao, B. Opposing effects of prednisolone treatment on T/NKT cell- and hepatotoxin-mediated hepatitis in mice. Hepatology 2014, 59, 1094–1106.
- Constant, P.; Davodeau, F.; Peyrat, M.A.; Poquet, Y.; Puzo, G.; Bonneville, M.; Fournié, J.J. Stimulation of human gamma delta T cells by nonpeptidic mycobacterial ligands. Science 1994, 264, 267–270.
- Kabelitz, D. Gamma Delta T Cells (γδ T Cells) in Health and Disease: In Memory of Professor Wendy Havran. Cells 2020, 9, 2564.
- Vantourout, P.; Hayday, A. Six-of-the-best: Unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 2013, 13, 88–100.
- Suzuki, T.; Minagawa, S.; Yamazaki, T.; Arai, T.; Kanai, M.; Shinjo, S.; Goda, N. Loss of hypoxia inducible factor-1α aggravates γδ T-cell-mediated inflammation during acetaminophen-induced liver injury. Hepatol. Commun. 2018, 2, 571–581.
- Ferrara, J.L.; Abhyankar, S.; Gilliland, D.G. Cytokine storm of graft-versus-host disease: A critical effector role for interleukin-1. Transpl. Proc. 1993, 25, 1216–1217.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851.
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273.
- Zhao, J.; Kim, J.W.; Zhou, Z.; Qi, J.; Tian, W.; Lim, C.W.; Han, K.M.; Kim, B. Macrophage-Inducible C-Type Lectin Signaling Exacer-bates Acetaminophen-Induced Liver Injury by Promoting Kupffer Cell Activation in Mice. Mol. Pharmacol. 2021, 99, 92–103.
- Campion, S.N.; Johnson, R.; Aleksunes, L.M.; Goedken, M.J.; van Rooijen, N.; Scheffer, G.L.; Cherrington, N.J.; Manautou, J.E. Hepatic Mrp4 induction following acetaminophen exposure is dependent on Kupffer cell function. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G294–G304.
- Liao, Y.; Yang, Y.; Wang, X.; Wei, M.; Guo, Q.; Zhao, L. Oroxyloside ameliorates acetaminophen-induced hepatotoxicity by inhibiting JNK related apoptosis and necroptosis. J. Ethnopharmacol. 2020, 258, 112917.
- Viswanathan, P.; Sharma, Y.; Jaber, F.L.; Tchaikovskaya, T.; Gupta, S. Transplanted hepatocytes rescue mice in acetaminophen-induced acute liver failure through paracrine signals for hepatic ATM and STAT3 pathways. FASEB J. 2021, 35, e21471.
- Jiang, W.P.; Deng, J.S.; Huang, S.S.; Wu, S.H.; Chen, C.C.; Liao, J.C.; Chen, H.Y.; Lin, H.Y.; Huang, G.J. Sanghuangporus sanghuangMyceli-um Prevents Paracetamol-Induced Hepatotoxicity through Regulating the MAPK/NF-κB, Keap1/Nrf2/HO-1, TLR4/PI3K/Akt, and CaMKKβ/LKB1/AMPK Pathways and Suppressing Oxidative Stress and Inflammation. Antioxidants 2021, 10, 897.
- Fisher, J.E.; McKenzie, T.J.; Lillegard, J.B.; Yu, Y.; Juskewitch, J.E.; Nedredal, G.I.; Brunn, G.J.; Yi, E.S.; Malhi, H.; Smyrk, T.C.; et al. Role of Kupffer cells and toll-like receptor 4 in acetaminophen-induced acute liver failure. J. Surg. Res. 2013, 180, 147–155.
- Bourdi, M.; Masubuchi, Y.; Reilly, T.P.; Amouzadeh, H.R.; Martin, J.L.; George, J.W.; Shah, A.G.; Pohl, L.R. Protection against aceta-minophen-induced liver injury and lethality by interleukin 10: Role of inducible nitric oxide synthase. Hepatology 2002, 35, 289–298.
- James, L.P.; Kurten, R.C.; Lamps, L.W.; McCullough, S.; Hinson, J.A. Tumour necrosis factor receptor 1 and hepatocyte regenera-tion in acetaminophen toxicity: A kinetic study of proliferating cell nuclear antigen and cytokine expression. Basic Clin. Pharma-Col. Toxicol. 2005, 97, 8–14.