 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patrick Legembre | -- | 1829 | 2022-07-21 14:52:44 | | | |
| 2 | Peter Tang | Meta information modification | 1829 | 2022-07-22 02:57:11 | | |
Video Upload Options
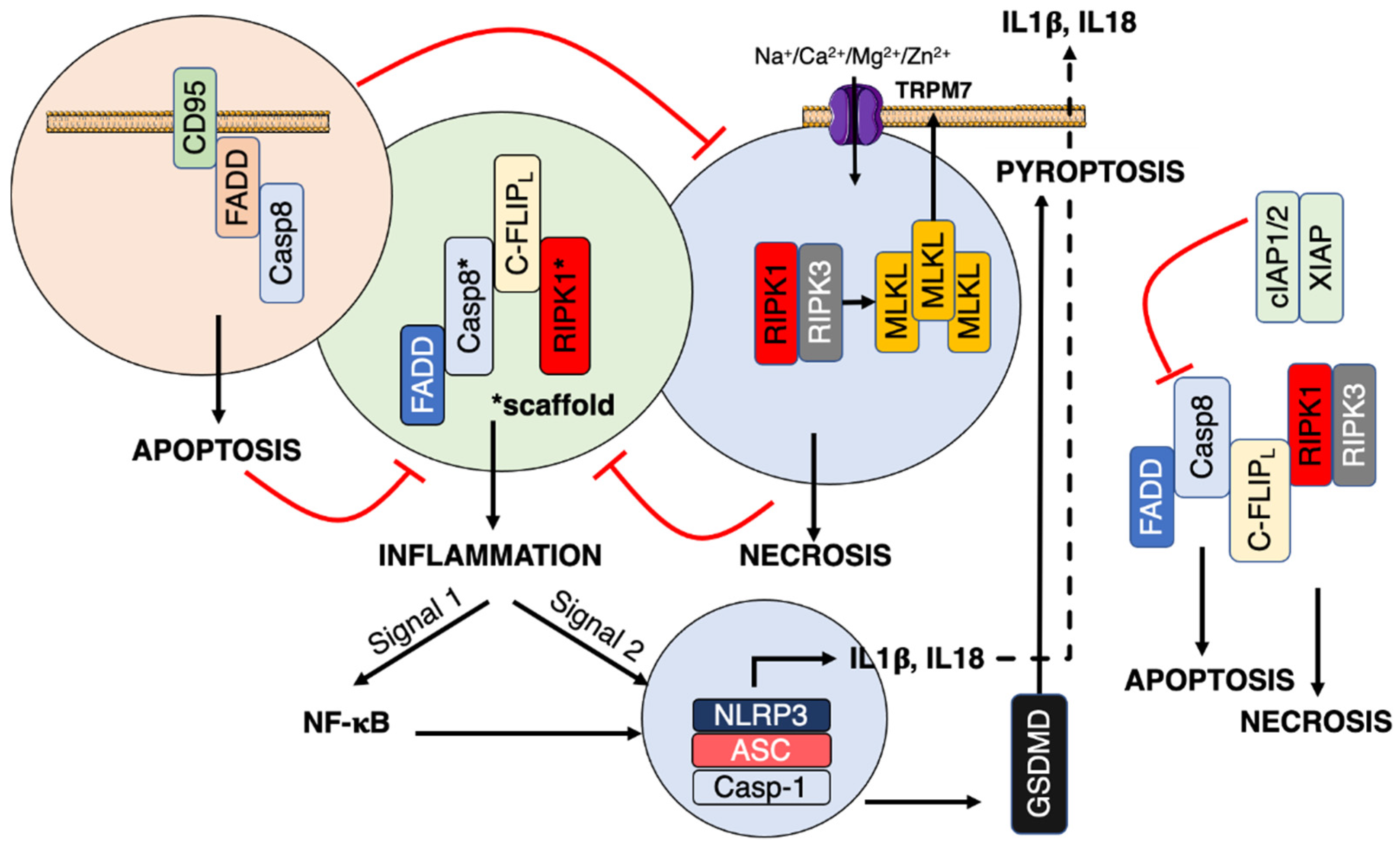
Study of the initial steps of the CD95-mediated signaling pathways is a field of intense research and a long list of actors has been described in the literature. Nonetheless, the dynamism of protein-protein interactions (PPIs) occurring in the presence or absence of its natural ligand, CD95L, and the cellular distribution where these PPIs take place render it difficult to predict what will be the cellular outcome associated with the receptor engagement. Accordingly, CD95 stimulation can trigger apoptosis, necroptosis, pyroptosis, or pro-inflammatory signaling pathways such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and phosphatidylinositol-3-kinase (PI3K). Recent data suggest that CD95 can also activate pattern recognition receptors (PRRs) known to sense damage-associated molecular patterns (DAMPs) such as DNA debris and dead cells. This activation might contribute to the pro-inflammatory role of CD95 and favor cancer development or severity of chronic inflammatory and auto-immune disorders.
1. Introduction
2. CD95 and DAMP-Sensor-Mediated Pathologies
2.1. Genetic Mutations in CD95 and DAMP Sensors and Chronic Inflammatory Diseases
2.1.1. CD95-Associated Genetic Disorders
|
ALPS Type |
Mutation Type |
|---|---|
|
ALPS-FAS |
CD95 germline homo- or heterozygous mutation |
|
ALPS-sFAS |
CD95 somatic mutation |
|
ALPS-FASLG |
CD95L germline mutation |
|
ALPS-CASP10 |
Caspase 10 germline mutations |
|
ALPS-U |
ALPS phenotype with no known-ALPS mutation |
2.1.2. DAMP-Sensor-Associated Genetic Disorders
|
DAMP-Sensor Mutation |
Mutation Type |
Autoinflammatory Disorder |
|---|---|---|
|
NLRP1 |
Loss of function |
NAIAD |
|
NLRP3 |
Gain of function |
CAPS = NOMID, FCAS, MWS |
|
NLRC4 |
Gain of function |
SCAN4, MAS |
|
Pyrin |
Gain of function of MEFV or loss of function of MVK |
FMF, HIDS, PAAND |
|
PSTPIP1 |
Loss of function |
PAPA |
3. CD95/Fas
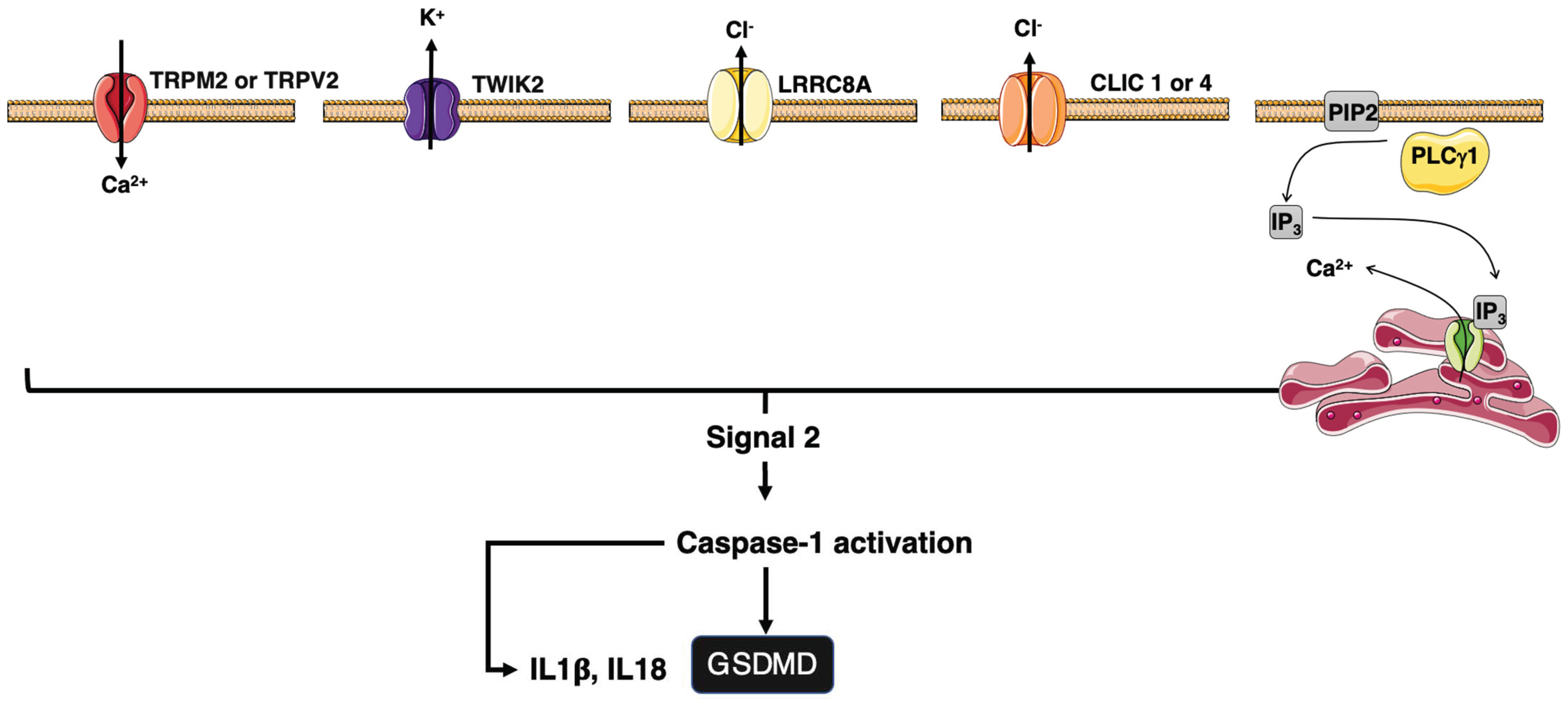
4. Inflammation
References
- Kurma, K.; Boizard-Moracchini, A.; Galli, G.; Jean, M.; Vacher, P.; Blanco, P.; Legembre, P. Soluble CD95L in cancers and chronic inflammatory disorders, a new therapeutic target? Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188596.
- Guegan, J.P.; Ginestier, C.; Charafe-Jauffret, E.; Ducret, T.; Quignard, J.F.; Vacher, P.; Legembre, P. CD95/Fas and metastatic disease: What does not kill you makes you stronger. Semin. Cancer Biol. 2019, 60, 121–131.
- Madkaikar, M.; Mhatre, S.; Gupta, M.; Ghosh, K. Advances in autoimmune lymphoproliferative syndromes. Eur. J. Haematol. 2011, 87, 1–9.
- Lenardo, M.J.; Oliveira, J.B.; Zheng, L.; Rao, V.K. ALPS-ten lessons from an international workshop on a genetic disease of apoptosis. Immunity 2010, 32, 291–295.
- Oliveira, J.B.; Bleesing, J.J.; Dianzani, U.; Fleisher, T.A.; Jaffe, E.S.; Lenardo, M.J.; Rieux-Laucat, F.; Siegel, R.M.; Su, H.C.; Teachey, D.T.; et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood 2010, 116, e35–e40.
- Li, P.; Huang, P.; Yang, Y.; Hao, M.; Peng, H.; Li, F. Updated Understanding of Autoimmune Lymphoproliferative Syndrome (ALPS). Clin. Rev. Allergy Immunol. 2016, 50, 55–63.
- Fields, M.L.; Sokol, C.L.; Eaton-Bassiri, A.; Seo, S.; Madaio, M.P.; Erikson, J. Fas/Fas ligand deficiency results in altered localization of anti-double-stranded DNA B cells and dendritic cells. J. Immunol. 2001, 167, 2370–2378.
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379.
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112.
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297.
- Krainer, J.; Siebenhandl, S.; Weinhausel, A. Systemic autoinflammatory diseases. J. Autoimmun. 2020, 109, 102421.
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Tran Mau-Them, F.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis. 2017, 76, 1191–1198.
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139.
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146.
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286.
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745.
- Moghaddas, F.; Llamas, R.; De Nardo, D.; Martinez-Banaclocha, H.; Martinez-Garcia, J.J.; Mesa-Del-Castillo, P.; Baker, P.J.; Gargallo, V.; Mensa-Vilaro, A.; Canna, S.; et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann. Rheum. Dis. 2017, 76, 2085–2094.
- Shoham, N.G.; Centola, M.; Mansfield, E.; Hull, K.M.; Wood, G.; Wise, C.A.; Kastner, D.L. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 13501–13506.
- Tummers, B.; Mari, L.; Guy, C.S.; Heckmann, B.L.; Rodriguez, D.A.; Ruhl, S.; Moretti, J.; Crawford, J.C.; Fitzgerald, P.; Kanneganti, T.D.; et al. Caspase-8-Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 2020, 52, 994–1006.e8.
- Siegel, R.M.; Frederiksen, J.K.; Zacharias, D.A.; Chan, F.K.-M.; Johnson, M.; Lynch, D.; Tsien, R.Y.; Lenardo, M.J. Fas Preassociation Required for Apoptosis Signaling and Dominant Inhibition by Pathogenic Mutations. Science 2000, 288, 2354–2357.
- Papoff, G.; Cascino, I.; Eramo, A.; Starace, G.; Lynch, D.H.; Ruberti, G. An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J. Immunol. 1996, 156, 4622–4630.
- Cascino, I.; Papoff, G.; De Maria, R.; Testi, R.; Ruberti, G. Fas/Apo-1 (CD95) receptor lacking the intracytoplasmic signaling domain protects tumor cells from Fas-mediated apoptosis. J. Immunol. 1996, 156, 13–17.
- Cascino, I.; Fiucci, G.; Papoff, G.; Ruberti, G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J. Immunol. 1995, 154, 2706–2713.
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588.
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160.
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem. 2006, 281, 13636–13643.
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257.
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703.
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700.
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327.
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227.
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65.
- Guegan, J.P.; Legembre, P. Nonapoptotic functions of Fas/CD95 in the immune response. FEBS J. 2018, 285, 809–827.
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367.
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372.
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526.
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Jin, C.; Flavell, R.A. Inflammasome activation. The missing link: How the inflammasome senses oxidative stress. Immunol. Cell Biol. 2010, 88, 510–512.


