Study of the initial steps of the CD95-mediated signaling pathways is a field of intense research and a long list of actors has been described in the literature. Nonetheless, the dynamism of protein-protein interactions (PPIs) occurring in the presence or absence of its natural ligand, CD95L, and the cellular distribution where these PPIs take place render it difficult to predict what will be the cellular outcome associated with the receptor engagement. Accordingly, CD95 stimulation can trigger apoptosis, necroptosis, pyroptosis, or pro-inflammatory signaling pathways such as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and phosphatidylinositol-3-kinase (PI3K). Recent data suggest that CD95 can also activate pattern recognition receptors (PRRs) known to sense damage-associated molecular patterns (DAMPs) such as DNA debris and dead cells. This activation might contribute to the pro-inflammatory role of CD95 and favor cancer development or severity of chronic inflammatory and auto-immune disorders.
- apoptosis
- auto-immunity
- CD95
- inflammasome
- necroptosis
- pyroptosis
- sting
1. Introduction
2. CD95 and DAMP-Sensor-Mediated Pathologies
2.1. Genetic Mutations in CD95 and DAMP Sensors and Chronic Inflammatory Diseases
2.1.1. CD95-Associated Genetic Disorders
In humans, CD95 mutation leads to the development of a disease called auto-immune lymphoproliferative syndrome or ALPS, also known as Canale–Smith syndrome [3]. ALPS involves the development of early onset polyclonal lymphoproliferation (splenomegaly, adenopathy) associated with expansion of a population of aberrant double-negative B220+CD3+TCRαβ+CD4−CD8− T cells. To qualify as ALPS, the evolution of the disease must be superior to 6 months and with exclusion of a secondary etiology for the lymphoproliferation (hematological malignancies or solid cancers, for example) [4,5][4][5]. This cardinal presentation is associated with mutation of CD95, CD95L, or caspase 10, a defect in lymphocyte proliferation, autoimmune cytopenia (mainly autoimmune hemolytic anemia and immune thrombocytopenic purpura), augmentation of Immunoglobulin A and G titers, associated with elevated dosage of B12 vitamin, augmentation of IL-10 or IL-18 and an increase in soluble-CD95L in patient serum. Of note, ALPS present an increased risk of hematological malignancies, which can complicate its diagnosis [6]. ALPS are divided into five subtypes, showing mutations in CD95, CD95L, or caspase-10 (see Table 1). Although most of the ALPS patients exhibit CD95 mutations, when Oliveira et al. revised the ALPS classification in 2010, lymphoproliferative disorders showing no mutations in CD95, CD95L, or caspase 10 were also classified as ALPS disorders (i.e., caspase 8, NRAS/KRAS, or SH2D1a mutations). In addition, Dianzani Autoimmune Lymphoproliferative Disease (DALD) was also reported as an ALPS-related disorder even though still no genetic defect was identified [4,5][4][5].ALPS Type | Mutation Type | ||||
|---|---|---|---|---|---|
ALPS-FAS | CD95 germline homo- or heterozygous mutation |
DAMP-Sensor Mutation | Mutation Type | Autoinflammatory Disorder | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
NLRP1 | Loss of function | NAIAD | ||||||||||||
ALPS-sFAS | CD95 somatic mutation | |||||||||||||
NLRP3 | Gain of function | CAPS = NOMID, FCAS, MWS | ALPS-FASLG | CD95L germline mutation | ||||||||||
ALPS-CASP10 | Caspase 10 germline mutations | |||||||||||||
ALPS-U | ALPS phenotype with no known-ALPS mutation |
2.1.2. DAMP-Sensor-Associated Genetic Disorders
DAMP are non-infectious triggers of the immune system released by dying or damaged cells [8] and detected by various membrane-bound or intracellular sensors, notably through a cytosolic complex called inflammasome [NLRC4 | Gain of function | SCAN4, MAS | ||||||
Pyrin | Gain of function of MEFV or loss of function of MVK | FMF, HIDS, PAAND | ||||||
PSTPIP1 | Loss of function | PAPA |
3. CD95/Fas
CD95 is a member of the tumor necrosis family receptors (TNF-Rs) and it is the prototype receptor to study the apoptotic signaling pathway. TNF-Rs are devoid of any enzymatic activity and necessitate protein-protein interactions (PPIs) to recruit enzymes such as proteases, kinases, and ubiquitin ligases to implement dynamic and complex signaling pathways. At the plasma membrane, CD95 auto-aggregates as homotrimer independently of its natural ligand, CD95L (also known as FasL or CD178) [20,21,22,23][20][21][22][23]. This trimeric structure is mandatory to implement cell death and rapidly forms larger signaling platforms in the presence of its natural ligand [20]. CD95 engagement induces the recruitment of the adaptor protein FADD, which in turn aggregates pro-caspase-8 in a complex-designated death-inducing signaling complex (DISC) [24]. Beyond DISC formation and induction of the apoptotic signal, FADD and caspase-8 contribute to different complexes involved in the induction of necroptosis or pyroptosis (discussed below). The switch between apoptosis and necroptosis has been discussed previously [25]. Briefly, ubiquitination of RIPK1 is a pivotal post-translational modification for the induction of the TNF-R1-mediated NF-κB activation [26,27][26][27] and its deubiquitination leads to the induction of cell death. The deubiquitinated RIPK1 is released from the death receptor and recruits TRADD, Fas-associated death domain (FADD), pro-caspase-8, and the long isoform of FLICE-like inhibitory protein (FLIPL) to trigger apoptosis [28]. In this complex, the caspase-8-mediated RIPK1 cleavage extinguishes the kinase activity. Degradation of c-IAP1 and c-IAP2 prevents the K63 ubiquitination of RIPK1 [29] and promotes the formation of another cellular complex in which FADD, pro-caspase-8, and FLIPL associate to trigger apoptosis. When caspase-8 is inactivated in these two complexes, the necrosome is formed. RIPK1 associates with RIPK3 to form this complex, in which RIPK3 phosphorylates MLKL to promote its plasma membrane distribution and the induction of necrosis [30,31,32][30][31][32]. Although CD95 engagement displays a broad range of cellular outcomes [33], whether such complexes occur in a CD95-dependent manner remains to be elucidated. Interestingly, the elimination of FADD or caspase-8 in mice does not lead to hyperplasia but instead is responsible for embryonic lethality, and these mice can survive post weaning when a double-ko mouse is realized with one of the necroptosis-mediating genes, RIPK3 [34,35][34][35] or MLKL [36] pointing out the tight control of the necroptotic machinery by the apoptotic one. Interestingly, caspase-8−/−/RIPK3−/− or caspase-8−/−/MLKL−/− double-ko mice develop hyperinflammation and lymphadenopathy, a phenotype resembling that observed in CD95-deficiency Lpr mice strongly suggesting that these factors control an additional mechanism in which CD95 could be involved too.4. Inflammation
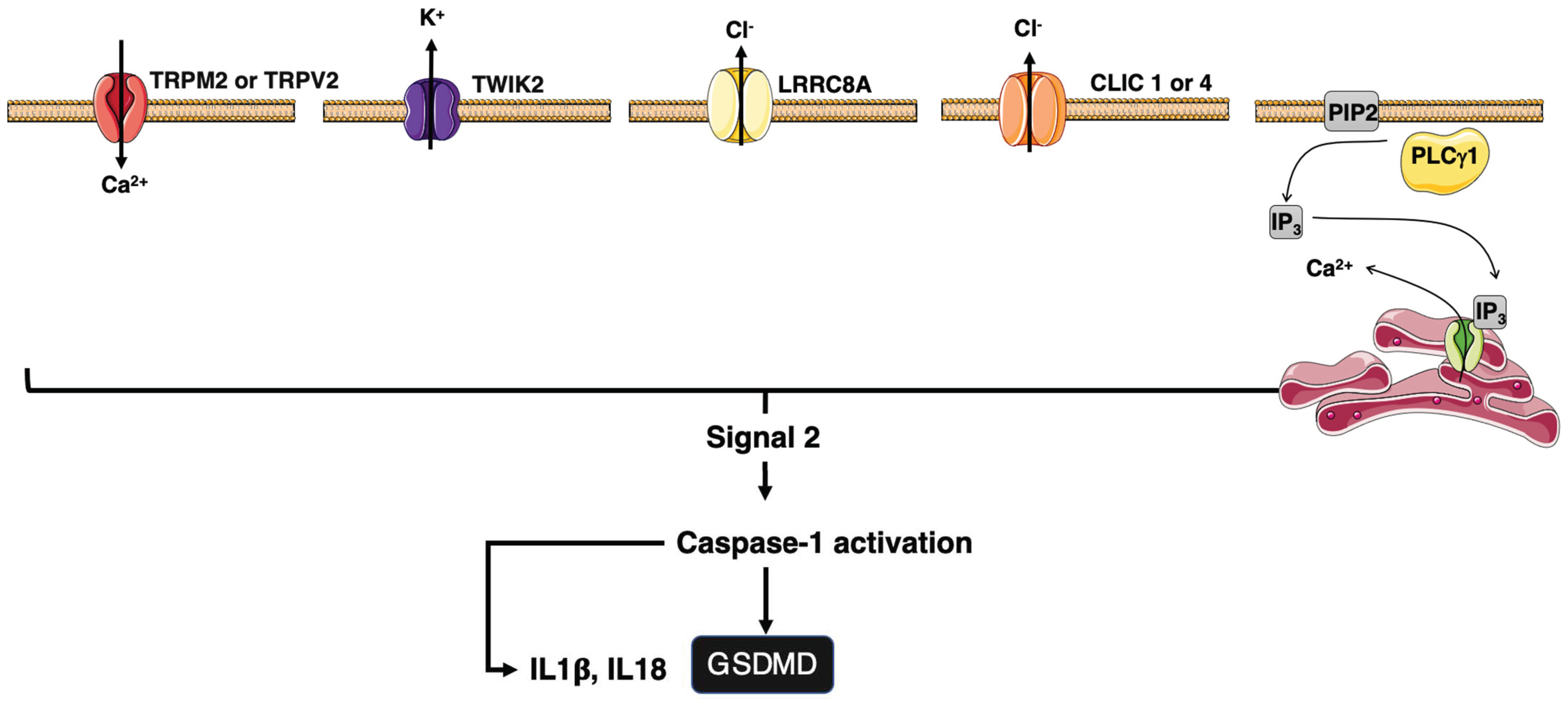
Inflammation after infection or injury relies on the detection of pathogen-associated and damage-associated molecular patterns (PAMPs/DAMPs). Examples of sterile DAMPs includes cholesterol crystals (atherosclerosis), β-amyloid (Alzheimer’s), islet amyloid polypeptide, ceramide, saturated fatty acids (type II diabetes), asbestos, silica dioxide (pulmonary fibrotic disorders), and monosodium urate (gout) [8,94][8][37]. Pattern recognition receptors (PRRs) translate the cell stress into proinflammatory signals. The cytosolic PAMPs/DAMPs activate inflammasome, which in turn induces caspase-1 activity leading to the maturation of interleukin-1β (IL-1β) and IL-18 [95,96][38][39]. Inflammasome can also induce pyroptosis by cleaving gasdermin D (GsdmD) [97,98][40][41]. GsdmD forms plasma membrane pores responsible for the induction of cell death and the release of mature IL-1β and IL-18 [99,100][42][43] (Figure 1). The signal 2 activating NLRP3 is diverse and includes mitochondrial reactive oxygen species (ROS), potassium efflux, and/or chloride influxes [101][44] (Figure 2), pointing out that NLRP3 is a sensor for a broad spectrum of cellular disturbances.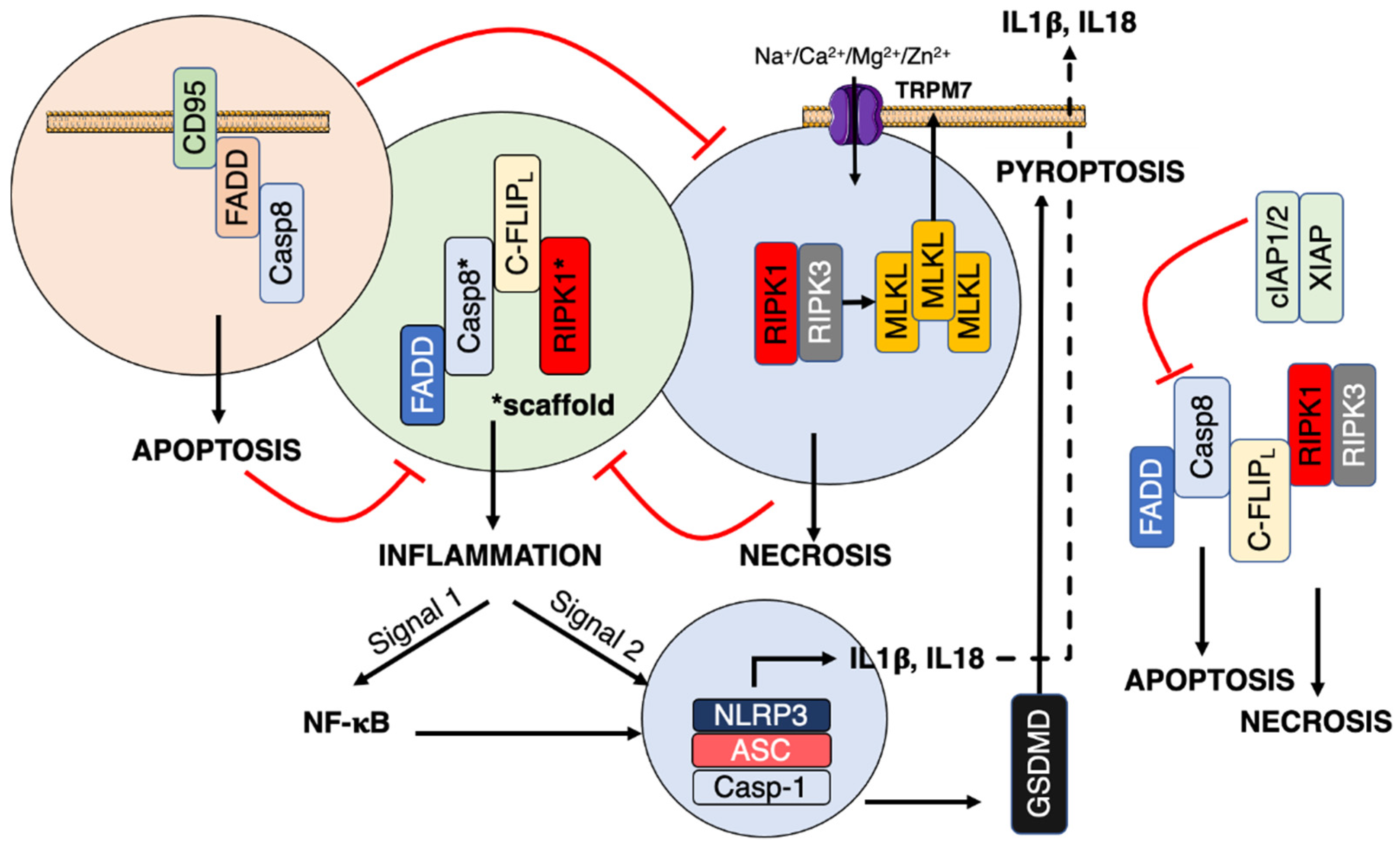
References
- Kurma, K.; Boizard-Moracchini, A.; Galli, G.; Jean, M.; Vacher, P.; Blanco, P.; Legembre, P. Soluble CD95L in cancers and chronic inflammatory disorders, a new therapeutic target? Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188596.
- Guegan, J.P.; Ginestier, C.; Charafe-Jauffret, E.; Ducret, T.; Quignard, J.F.; Vacher, P.; Legembre, P. CD95/Fas and metastatic disease: What does not kill you makes you stronger. Semin. Cancer Biol. 2019, 60, 121–131.
- Madkaikar, M.; Mhatre, S.; Gupta, M.; Ghosh, K. Advances in autoimmune lymphoproliferative syndromes. Eur. J. Haematol. 2011, 87, 1–9.
- Lenardo, M.J.; Oliveira, J.B.; Zheng, L.; Rao, V.K. ALPS-ten lessons from an international workshop on a genetic disease of apoptosis. Immunity 2010, 32, 291–295.
- Oliveira, J.B.; Bleesing, J.J.; Dianzani, U.; Fleisher, T.A.; Jaffe, E.S.; Lenardo, M.J.; Rieux-Laucat, F.; Siegel, R.M.; Su, H.C.; Teachey, D.T.; et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood 2010, 116, e35–e40.
- Li, P.; Huang, P.; Yang, Y.; Hao, M.; Peng, H.; Li, F. Updated Understanding of Autoimmune Lymphoproliferative Syndrome (ALPS). Clin. Rev. Allergy Immunol. 2016, 50, 55–63.
- Fields, M.L.; Sokol, C.L.; Eaton-Bassiri, A.; Seo, S.; Madaio, M.P.; Erikson, J. Fas/Fas ligand deficiency results in altered localization of anti-double-stranded DNA B cells and dendritic cells. J. Immunol. 2001, 167, 2370–2378.
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379.
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112.
- McGonagle, D.; McDermott, M.F. A proposed classification of the immunological diseases. PLoS Med. 2006, 3, e297.
- Krainer, J.; Siebenhandl, S.; Weinhausel, A. Systemic autoinflammatory diseases. J. Autoimmun. 2020, 109, 102421.
- Grandemange, S.; Sanchez, E.; Louis-Plence, P.; Tran Mau-Them, F.; Bessis, D.; Coubes, C.; Frouin, E.; Seyger, M.; Girard, M.; Puechberty, J.; et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann. Rheum. Dis. 2017, 76, 1191–1198.
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139.
- Canna, S.W.; de Jesus, A.A.; Gouni, S.; Brooks, S.R.; Marrero, B.; Liu, Y.; DiMattia, M.A.; Zaal, K.J.; Sanchez, G.A.; Kim, H.; et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat. Genet. 2014, 46, 1140–1146.
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286.
- Schnappauf, O.; Chae, J.J.; Kastner, D.L.; Aksentijevich, I. The Pyrin Inflammasome in Health and Disease. Front. Immunol. 2019, 10, 1745.
- Moghaddas, F.; Llamas, R.; De Nardo, D.; Martinez-Banaclocha, H.; Martinez-Garcia, J.J.; Mesa-Del-Castillo, P.; Baker, P.J.; Gargallo, V.; Mensa-Vilaro, A.; Canna, S.; et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann. Rheum. Dis. 2017, 76, 2085–2094.
- Shoham, N.G.; Centola, M.; Mansfield, E.; Hull, K.M.; Wood, G.; Wise, C.A.; Kastner, D.L. Pyrin binds the PSTPIP1/CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 13501–13506.
- Tummers, B.; Mari, L.; Guy, C.S.; Heckmann, B.L.; Rodriguez, D.A.; Ruhl, S.; Moretti, J.; Crawford, J.C.; Fitzgerald, P.; Kanneganti, T.D.; et al. Caspase-8-Dependent Inflammatory Responses Are Controlled by Its Adaptor, FADD, and Necroptosis. Immunity 2020, 52, 994–1006.e8.
- Siegel, R.M.; Frederiksen, J.K.; Zacharias, D.A.; Chan, F.K.-M.; Johnson, M.; Lynch, D.; Tsien, R.Y.; Lenardo, M.J. Fas Preassociation Required for Apoptosis Signaling and Dominant Inhibition by Pathogenic Mutations. Science 2000, 288, 2354–2357.
- Papoff, G.; Cascino, I.; Eramo, A.; Starace, G.; Lynch, D.H.; Ruberti, G. An N-terminal domain shared by Fas/Apo-1 (CD95) soluble variants prevents cell death in vitro. J. Immunol. 1996, 156, 4622–4630.
- Cascino, I.; Papoff, G.; De Maria, R.; Testi, R.; Ruberti, G. Fas/Apo-1 (CD95) receptor lacking the intracytoplasmic signaling domain protects tumor cells from Fas-mediated apoptosis. J. Immunol. 1996, 156, 13–17.
- Cascino, I.; Fiucci, G.; Papoff, G.; Ruberti, G. Three functional soluble forms of the human apoptosis-inducing Fas molecule are produced by alternative splicing. J. Immunol. 1995, 154, 2706–2713.
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588.
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160.
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem. 2006, 281, 13636–13643.
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257.
- Wang, L.; Du, F.; Wang, X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell 2008, 133, 693–703.
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700.
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327.
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227.
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65.
- Guegan, J.P.; Legembre, P. Nonapoptotic functions of Fas/CD95 in the immune response. FEBS J. 2018, 285, 809–827.
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367.
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–372.
- Alvarez-Diaz, S.; Dillon, C.P.; Lalaoui, N.; Tanzer, M.C.; Rodriguez, D.A.; Lin, A.; Lebois, M.; Hakem, R.; Josefsson, E.C.; O’Reilly, L.A.; et al. The Pseudokinase MLKL and the Kinase RIPK3 Have Distinct Roles in Autoimmune Disease Caused by Loss of Death-Receptor-Induced Apoptosis. Immunity 2016, 45, 513–526.
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342.
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426.
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Jin, C.; Flavell, R.A. Inflammasome activation. The missing link: How the inflammasome senses oxidative stress. Immunol. Cell Biol. 2010, 88, 510–512.