Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anabela Araújo Ferreira | -- | 1810 | 2022-07-19 23:11:41 | | | |
| 2 | Dean Liu | Meta information modification | 1810 | 2022-07-21 03:41:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ferreira, A.; Pereira, F.; Reis, C.; Oliveira, M.J.; Sousa, M.J.; Preto, A. Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation. Encyclopedia. Available online: https://encyclopedia.pub/entry/25353 (accessed on 28 July 2026).
Ferreira A, Pereira F, Reis C, Oliveira MJ, Sousa MJ, Preto A. Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation. Encyclopedia. Available at: https://encyclopedia.pub/entry/25353. Accessed July 28, 2026.
Ferreira, Anabela, Flávia Pereira, Celso Reis, Maria José Oliveira, Maria João Sousa, Ana Preto. "Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation" Encyclopedia, https://encyclopedia.pub/entry/25353 (accessed July 28, 2026).
Ferreira, A., Pereira, F., Reis, C., Oliveira, M.J., Sousa, M.J., & Preto, A. (2022, July 20). Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation. In Encyclopedia. https://encyclopedia.pub/entry/25353
Ferreira, Anabela, et al. "Oncogenic KRAS Mutations in Apoptosis and Autophagy Regulation." Encyclopedia. Web. 20 July, 2022.
Copy Citation
KRAS, one of the RAS protein family members, plays an important role in autophagy and apoptosis, through the regulation of several downstream effectors. In cancer cells, KRAS mutations confer the constitutive activation of this oncogene, stimulating cell proliferation, inducing autophagy, suppressing apoptosis, altering cell metabolism, changing cell motility and invasion and modulating the tumor microenvironment.
KRAS mutations
cell death resistance
apoptosis
autophagy
1. Introduction
RAS proteins are a family of small monomeric guanosine triphosphatases (GTPases) that function as transducers of extracellular stimuli to intracellular signaling. RAS proteins regulate important cellular functions, including apoptosis, autophagy, cell proliferation, differentiation, gene expression, migration, invasion and tumor microenvironment (TME) [1][2][3][4][5].
The RAS family includes Harvey (H-)RAS, Kirsten (K-)RAS and Neuroblastoma (N-) RAS genes, which are the most frequent oncogenes and one of the most prevalent drivers of cancer. These three RAS genes encode four homologous proteins: HRAS, NRAS, KRAS4A and KRAS4B, whose structures and sequences are highly conserved. RAS isoforms share 85–90% sequence homology in the G-domain and diverge mainly at the C-terminal [6][7][8]. The G-domain contains G motifs, which bind directly to GDP or GTP, such as switch I and II and the P-loop. The C-terminal disparities are caused by post-translational modifications (PTMs) that specifically occur in a string of residues termed the hypervariable region (HVR), which is responsible for appropriate membrane localization, interaction and cellular trafficking of the proteins. Therefore, such C-terminal disparities result in different subcellular localization, that can be linked to variations in the diversity or amplitude of signaling [8][9]. In fact, KRAS has a polybasic region required for plasma membrane localization, whereas HRAS and NRAS are palmitoylated; therefore, they are more likely to localize into lipid rafts [9][10]. KRAS4A and KRAS4B are protein products generated through alternative gene splicing of the fourth exon of the KRAS gene, which determines the presence or absence of exon 4A. The alternative fourth exon encodes the HVRs responsible for membrane targeting. Thus, KRAS4A is palmitoylated, whereas KRAS4B is not, because it lacks a site of palmitoylation [11]. Additionally, KRAS4A is expressed at low levels, whereas KRAS4B (hereafter referred to as KRAS) is ubiquitously expressed and accounts for 90–99% of all KRAS mRNA forms [12][13].
Despite their high homology, the functions of RAS proteins also differ significantly and do not display redundant roles. This is particularly surprising since the regions of the proteins that interact with downstream effectors are identical to the three RAS isoforms. Therefore, their specific roles may be explained by various others factors, such as cellular context, differential interaction with effectors, compartmentalized signaling and PTMs [14].
Regarding oncogenic RAS mutations, they are found in approximately 30% of all human cancers and contribute to important aspects of the malignant phenotype, such as invasion, programmed cell death, deregulation of tumor-cell growth and the induction of new blood-vessels’ formation [3][15][16]. From all human cancers, RAS mutations are more frequently found in about 60–90% of pancreatic cancer cases, followed by approximately 30–50% of colorectal cancer (CRC) cases, and between a range of 20% and 30% of lung cancer cases [12][17].
2. The Oncogene KRAS
As a member of the human RAS family, the KRAS oncogene encodes a 21 kDa small GTPase, which functions as an on/off switch protein that alternates between an active GTP-bound and an inactive GDP-bound state, by cleaving the terminal phosphate of the nucleotide. The on/off state of KRAS is regulated by GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) (Figure 1). GAPs have a GTPase activity responsible for the inactivation of KRAS, through the hydrolysis of GTP. In turn, GEFs facilitate the activation of KRAS, forcing the release of bound GDP and allowing its replacement by GTP. In mammalian cells, KRAS is normally found in its inactive state—GDP-bound [4][18][19].
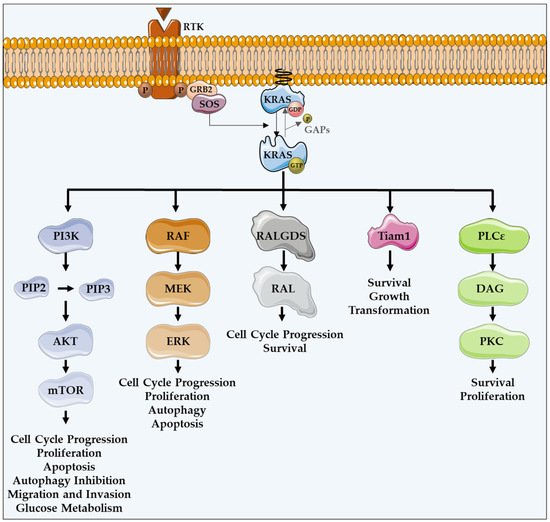
Figure 1. KRAS major effectors and cellular functions. KRAS proteins are activated upon activation of an RTK-like EGFR. KRAS is a GTPase that functions as an on/off switch that alternates between an active GTP-bound and an inactive GDP-bound state, regulated by GAPs and GEFs, such as Sos1. This oncogene regulates several effector pathways, including the MAPK and PI3K/AKT pathways. GTP-bound KRAS leads to the activation of RAF proteins, resulting in the initiation of MAPK signaling. Subsequently, MEK is activated and, in turn, phosphorylates and activates ERK. Downstream of this, ERK can regulate numerous transcription factors, promoting cell cycle progression and influencing proliferation and apoptosis. Besides MAPK pathway, KRAS can interact with PI3K, whose activation leads to the phosphorylation of PIP2, resulting in PIP3. This second messenger is able to activate a large number of proteins containing a pleckstrin homology domain, including PDK1 and AKT, whose main downstream effector is mTOR. These proteins regulate cell cycle progression, cell survival, glucose metabolism, cell growth and proliferation. In addition to MAPK and PI3K/AKT pathways, KRAS can activate RALGDS, whose downstream effector is RAL GTPases, promoting cell cycle progression and survival. Furthermore, KRAS interacts with Tiam1, a Rho family GTPase, which is implicated in the development of RAS-driven tumors, growth transformation and promotion of cell survival. KRAS also binds to PLCε 44, a phospho-lipase C isoform responsible for KRAS mediated production of DAG, resulting in calcium release and activation of the PKC signaling cascade, involved in survival, proliferation, and calcium mobilization.
KRAS proteins are activated following the activation of different receptor tyrosine kinases (RTK), such as the epidermal growth factor receptor (EGFR) [12][16]. Upon ligand binding in the extracellular portion, RTKs dimerize, resulting in conformational changes that lead to the autophosphorylation of their intracellular carboxyl-terminal domain. Such phosphorylation stimulates the binding of proteins containing SH2 domains, also known as docking adaptor proteins, to the phosphorylated tyrosine residues, turning RTKs able to recruit GEFs. Grb2 is an example of an adaptor molecule that binds directly to RTKs, or through other adaptor proteins present on growth factor receptors, such as IRS. Grb2 associates to Sos1, which is a GEF, activating the KRAS-RAF-MEK-ERK-MAPK pathway [20][21]. GEFs interact and activate KRAS, promoting the dissociation of GDP and the binding of GTP [22][23][24]. KRAS, in a GTP-bound active state, transduces intracellular signals through other GTPases and kinases, thus linking the presence of extracellular growth factors to intracellular signaling cascades. There are several intracellular signaling cascades activated by KRAS (Figure 1) [25].
Once they are active, KRAS proteins transduce signals across the plasma membrane [12][16], stimulating several effectors by the recruitment and activation of proteins involved in the propagation of signaling from growth factors and other receptors [18][26].
The protein serine/threonine kinase rapidly-accelerated fibrosarcoma (RAF) was the first RAS effector to be characterized and is still the best known. Upon binding to KRAS-GTP, RAF proteins are relocated to the plasma membrane and activated, leading to the initiation of the mitogen-associated protein kinase (MAPK) cascade. This kinase phosphorylates and activates MAPK/ERK kinase (MEK), which subsequently phosphorylates and activates extracellular signal-regulated kinase (ERK). Downstream of this, ERK can regulate numerous transcription factors and cellular functions, such as cell cycle progression, proliferation, autophagy and apoptosis (Figure 1) [16][27].
In addition to the MAPK pathway, KRAS can interact with phosphatidylinositol 3-kinase (PI3K), whose activation leads to the phosphorylation of phosphatidylinositol-4,5-diphosphate (PIP2), resulting in the phosphatidylinositol-3,4,5-triphosphate (PIP3) production. This second messenger is able to recruit and activate a large number of proteins containing a pleckstrin homology domain, including phosphatidylinositol dependent kinase 1 (PDK1) and AKT, whose main downstream effector is a mammalian target of rapamycin (mTOR). Such proteins transmit signals regulating cell cycle progression, proliferation, apoptosis, autophagy, migration, invasion and glucose metabolism (Figure 1) [12][16][26][28].
Another important KRAS effector is RAL guanine nucleotide dissociation stimulator (RALGDS), which activates RAS-like (RAL) GTPases and has pro-survival functions and promotes the cell cycle progression. Additionally, KRAS interacts with T-lymphoma invasion and metastasis protein 1 (Tiam1), a Rho family GTPase, which is implicated in the development of RAS-driven tumors, growth transformation, promotion of cell survival, activation of the c-Jun amino-terminal kinase (JNK) mitogen-activated protein kinase and the activation of the NF-kB transcription factor [29]. RAS also binds to PLCε 44, a phospholipase C isoform responsible for the RAS-mediated production of the membrane lipid diacylglycerol (DAG), which results in calcium release and activation of the PKC signaling cascade involved in survival, proliferation, and calcium mobilization (Figure 1) [12][16][29][30].
The combined action of these signaling pathways regulated by this oncogene can lead to several features of malignant transformation, if cells express KRAS-activated mutants [16].
3. KRAS Mutations and Cancer
Of the three RAS isoforms, KRAS has the highest mutation rate (86%) and leads to a poor prognosis. Oncogenic KRAS mutations are a hallmark of cancer, being a very frequent event in many cancers, including pancreatic cancers (90%), CRCs (30–50%) and lung cancers, especially non-small-cell lung cancer (NSCLC) (15–20%). Such mutations are also present in endometrial cancer, biliary tract malignancies, cervical cancer, liver cancer, bladder cancer, breast cancer and myeloid leukemia [31][32][33].
KRAS mutations, featured by single base missense mutations, lead to alterations in the homeostatic balance of GTP and GDP binding, resulting in its constitutively GTP-bound active state, through the reduction in GTP hydrolysis or the increase in the rate of GTP loading. Thus, mutated KRAS is able to constitutively activate oncogenic pathways and cellular signal transduction [7][34][35][36]. Point substitutions in codons 12 and 13 are the most common oncogenic KRAS mutations, representing 90% of them. In addition, mutations occur less frequently in codons 61, 63, 117, 119 and 146 [32][37]. In detail, the hotspot KRAS-mutated codons 12 and 13 correspond to a glycine, and are positioned in the P-loop of KRAS protein, which is essential in maintaining its active form. In these codons, the replacement of glycine by other amino acids, except proline, prevents the arginine finger of GAPs from promoting hydrolysis of GTP [14][21][34][38][39]. Thus, the KRAS hotspot mutations result in insensitivity to GAPs increasing time in the GTP bound state [21][22][40][41][42][43][44][45]. In other words, these KRAS mutations prevent GAPs from promoting GTP hydrolysis, resulting in the constitutive activation of the KRAS protein and the downstream pathways [24][41][45][46]. Different amino acid substitutions activate different KRAS downstream signaling pathways and display different clonogenic growth potential and responses to targeted therapies. This happens because different mutations influence the way the interaction between KRAS and its effectors occurs [21][45]. Codon 12 mutations increase aggressiveness by the differential regulation of KRAS downstream pathways that leads to the inhibition of apoptosis, the enhanced loss of contact inhibition, and the increased predisposition to anchorage-independent growth. Codon 13 mutations lead to reduced transforming capacity compared to codon 12 mutations [43]. Alternatively, codon 61 substitutions activate KRAS through a similar mechanism, indicating the essential nature of codon 61 in KRAS deactivation [21]. This constitutively active KRAS protein contributes to cell proliferation, suppression of apoptosis, altered cell metabolism and changes in the tumor microenvironment, which leads to tumorigenesis, tumor maintenance, invasion and metastasis [2][24][46][47].
Generally, tumors harboring KRAS mutations present higher resistance to chemotherapy and EGFR-inhibitors-targeted therapy, including cetuximab and panitumumab, leading to a worse overall survival, especially in CRC [37][48].
In addition, the direct inhibition of KRAS has not presented successful results and efforts have been made to focus on targeting their downstream signaling proteins [49]. This is why it is so important to find the Achilles heel of mutated KRAS. KRAS is implicated in the regulation of crucial cellular processes that can prevent tumorigenesis, including apoptosis and autophagy [1][12]. However, there are still several unanswered questions regarding the role of KRAS mutations in autophagy and apoptosis regulation, and their regulation loop.
References
- Alves, S.; Castro, L.; Fernandes, M.S.; Francisco, R.; Castro, P.; Priault, M.; Chaves, S.R.; Moyer, M.P.; Oliveira, C.; Seruca, R.; et al. Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 2015, 6, 30787–30802.
- Smith, G.; Bounds, R.; Wolf, H.; Steele, R.; Carey, F.; Wolf, C. Activating K-Ras mutations outwith ‘hotspot’ codons in sporadic colorectal tumours—implications for personalised cancer medicine. Br. J. Cancer 2010, 102, 693–703.
- Leung, E.L.H.; Luo, L.X.; Liu, Z.Q.; Wong, V.K.W.; Lu, L.L.; Xie, Y.; Zhang, N.; Qu, Y.Q.; Fan, X.X.; Li, Y.; et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: Blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018, 9, 216.
- Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and predictive roles of KRAS mutation in colorectal cancer. Int. J. Mol. Sci. 2012, 13, 12153–12168.
- Liu, X.; Jakubowski, M.; Hunt, J.L. KRAS gene mutation in colorectal cancer is correlated with increased proliferation and spontaneous apoptosis. Am. J. Clin. Pathol. 2011, 135, 245–252.
- Overmeyer, J.H.; Maltese, W.A. Death pathways triggered by activated Ras in cancer cells. Front. Biosci. (Landmark Ed.) 2011, 16, 1693–1713.
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small GTPases 2010, 1, 2–27.
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384.
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51.
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39.
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784.
- Cazzanelli, G.; Pereira, F.; Alves, S.; Francisco, R.; Azevedo, L.; Dias Carvalho, P.; Almeida, A.; Côrte-Real, M.; Oliveira, M.J.; Lucas, C.; et al. The Yeast Saccharomyces Cerevisiae as a Model for Understanding RAS Proteins and Their Role in Human Tumorigenesis. Cells 2018, 7, 14.
- Wang, Y.; You, M.; Wang, Y. Alternative splicing of the K-ras gene in mouse tissues and cell lines. Exp. Lung Res. 2001, 27, 255–267.
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2468.
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116, 6607–6665.
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22.
- Yoo, B.H.; Khan, I.A.; Koomson, A.; Gowda, P.; Sasazuki, T.; Shirasawa, S.; Gujar, S.; Rosen, K.V. Oncogenic RAS-induced downregulation of ATG12 is required for survival of malignant intestinal epithelial cells. Autophagy 2018, 14, 134–151.
- Xu, K.; Park, D.; Magis, A.T.; Zhang, J.; Zhou, W.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Deng, X. Small Molecule KRAS Agonist for Mutant KRAS Cancer Therapy. Mol. Cancer 2019, 18, 85.
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142.
- Fröjdö, S.; Vidal, H.; Pirola, L. Alterations of insulin signaling in type 2 diabetes: A review of the current evidence from humans. Biochim. Biophys. Acta—Mol. Basis Dis. 2009, 1792, 83–92.
- Ihle, N. Differential Activity of the KRAS Oncogene by Method of Activation: Implications for Signaling and Therapeutic Intervention. Ph.D. Thesis, The University of Texas Graduate School of Biomedical Sciences, Houston, TX, USA, 2012.
- Sancho, E.; Batlle, E.; Clevers, H. Signaling Pathways in Intestinal Development and Cancer. Annu. Rev. Cell Dev. Biol. 2004, 20, 695–723.
- Levy, R.; Biran, A.; Poirier, F.; Raz, A.; Kloog, Y. Galectin-3 mediates cross-talk between K-ras and let-7c tumor suppressor microRNA. PLoS ONE 2011, 6, e27490.
- Knickelbein, K.; Zhang, L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015, 2, 4–12.
- Martinelli, E.; De Palma, R.; Orditura, M.; De Vita, F.; Ciardiello, F. Anti-epidermal growth factor receptor monoclonal antibodies in cancer therapy. Clin. Exp. Immunol. 2009, 158, 1–9.
- Okudela, K.; Hayashi, H.; Ito, T.; Yazawa, T.; Suzuki, T.; Nakane, Y.; Sato, H.; Ishi, H.; KeQin, X.; Masuda, A.; et al. K-ras Gene Mutation Enhances Motility of Immortalized Airway Cells and Lung Adenocarcinoma Cells via Akt Activation: Possible Contribution to Non-Invasive Expansion of Lung Adenocarcinoma. Am. J. Pathol. 2004, 164, 91–100.
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465.
- Efferth, T. Signal Transduction Pathways of the Epidermal Growth Factor Receptor in Colorectal Cancer and their Inhibition by Small Molecules. Curr. Med. Chem. 2012, 19, 5735–5744.
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625.
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, Š. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195.
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol. Cancer 2010, 9, 293.
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879.
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell Rep. Med. 2021, 2, 100186.
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697.
- Scheffzek, K.; Lautwein, A.; Kabsch, W.; Ahmadian, M.R.; Wittinghofer, A. Crystal structure of the GTPase-activating domain of human p120GAP and implications for the interaction with Ras. Nature 1996, 384, 591–596.
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic ras mutants. Science 1997, 277, 333–338.
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 2010, 150960.
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33.
- Fan, G.; Lou, L.; Song, Z.; Zhang, X.; Xiong, X.F. Targeting mutated GTPase KRAS in tumor therapies. Eur. J. Med. Chem. 2021, 226, 113816.
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072.
- Worthley, D.-L.; Leggett, B.-A. Colorectal cancer: Molecular features and clinical opportunities. Clin. Biochem. Rev. 2010, 31, 31–38.
- Krasinskas, A.M. EGFR Signaling in Colorectal Carcinoma. Patholog. Res. Int. 2011, 2011, 932932.
- Bruera, G.; Cannita, K.; Di Giacomo, D.; Lamy, A.; Frébourg, T.; Sabourin, J.C.; Tosi, M.; Alesse, E.; Ficorella, C.; Ricevuto, E. Worse prognosis of KRAS c.35 G > A mutant metastatic colorectal cancer (MCRC) patients treated with intensive triplet chemotherapy plus bevacizumab (FIr-B/FOx). BMC Med. 2013, 11, 59.
- Roper, J.; Hung, K.E. Molecular Mechanisms of Colorectal Carcinogenesis. In Molecular Pathogenesis of Colorectal Cancer; Haigis, K.M., Ed.; Springer: New York, NY, USA, 2013; pp. 25–66. ISBN 978-1-4614-8411-0.
- Hammond, D.E.; Mageean, C.J.; Rusilowicz, E.V.; Wickenden, J.A.; Clague, M.J.; Prior, I.A. Differential Reprogramming of Isogenic Colorectal Cancer Cells by Distinct Activating KRAS Mutations. J. Proteome Res. 2015, 14, 1535–1546.
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386.
- Levy, R.; Grafi-Cohen, M.; Kraiem, Z.; Kloog, Y. Galectin-3 Promotes Chronic Activation of K-Ras and Differentiation Block in Malignant Thyroid Carcinomas. Am. Assoc. Cancer Res. 2010, 9, 2208–2219.
- Sideris, M.; Emin, E.I.; Abdullah, Z.; Hanrahan, J.; Stefatou, K.M.; Sevas, V.; Emin, E.; Hollingworth, T.; Odejinmi, F.; Papagrigoriadis, S.; et al. The role of KRAS in endometrial cancer: A mini-review. Anticancer Res. 2019, 39, 533–540.
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851.
More
Information
Subjects:
Cell Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
21 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No