Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Anabela Araújo Ferreira and Version 2 by Dean Liu.
KRAS, one of the RAS protein family members, plays an important role in autophagy and apoptosis, through the regulation of several downstream effectors. In cancer cells, KRAS mutations confer the constitutive activation of this oncogene, stimulating cell proliferation, inducing autophagy, suppressing apoptosis, altering cell metabolism, changing cell motility and invasion and modulating the tumor microenvironment.
- KRAS mutations
- cell death resistance
- apoptosis
- autophagy
1. Introduction
RAS proteins are a family of small monomeric guanosine triphosphatases (GTPases) that function as transducers of extracellular stimuli to intracellular signaling. RAS proteins regulate important cellular functions, including apoptosis, autophagy, cell proliferation, differentiation, gene expression, migration, invasion and tumor microenvironment (TME) [1][2][3][4][5][1,2,3,4,5].
The RAS family includes Harvey (H-)RAS, Kirsten (K-)RAS and Neuroblastoma (N-) RAS genes, which are the most frequent oncogenes and one of the most prevalent drivers of cancer. These three RAS genes encode four homologous proteins: HRAS, NRAS, KRAS4A and KRAS4B, whose structures and sequences are highly conserved. RAS isoforms share 85–90% sequence homology in the G-domain and diverge mainly at the C-terminal [6][7][8][6,7,8]. The G-domain contains G motifs, which bind directly to GDP or GTP, such as switch I and II and the P-loop. The C-terminal disparities are caused by post-translational modifications (PTMs) that specifically occur in a string of residues termed the hypervariable region (HVR), which is responsible for appropriate membrane localization, interaction and cellular trafficking of the proteins. Therefore, such C-terminal disparities result in different subcellular localization, that can be linked to variations in the diversity or amplitude of signaling [8][9][8,9]. In fact, KRAS has a polybasic region required for plasma membrane localization, whereas HRAS and NRAS are palmitoylated; therefore, they are more likely to localize into lipid rafts [9][10][9,10]. KRAS4A and KRAS4B are protein products generated through alternative gene splicing of the fourth exon of the KRAS gene, which determines the presence or absence of exon 4A. The alternative fourth exon encodes the HVRs responsible for membrane targeting. Thus, KRAS4A is palmitoylated, whereas KRAS4B is not, because it lacks a site of palmitoylation [11]. Additionally, KRAS4A is expressed at low levels, whereas KRAS4B (hereafter referred to as KRAS) is ubiquitously expressed and accounts for 90–99% of all KRAS mRNA forms [12][13][12,13].
Despite their high homology, the functions of RAS proteins also differ significantly and do not display redundant roles. This is particularly surprising since the regions of the proteins that interact with downstream effectors are identical to the three RAS isoforms. Therefore, their specific roles may be explained by various others factors, such as cellular context, differential interaction with effectors, compartmentalized signaling and PTMs [14].
Regarding oncogenic RAS mutations, they are found in approximately 30% of all human cancers and contribute to important aspects of the malignant phenotype, such as invasion, programmed cell death, deregulation of tumor-cell growth and the induction of new blood-vessels’ formation [3][15][16][3,15,16]. From all human cancers, RAS mutations are more frequently found in about 60–90% of pancreatic cancer cases, followed by approximately 30–50% of colorectal cancer (CRC) cases, and between a range of 20% and 30% of lung cancer cases [12][17][12,17].
2. The Oncogene KRAS
As a member of the human RAS family, the KRAS oncogene encodes a 21 kDa small GTPase, which functions as an on/off switch protein that alternates between an active GTP-bound and an inactive GDP-bound state, by cleaving the terminal phosphate of the nucleotide. The on/off state of KRAS is regulated by GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs) (Figure 1). GAPs have a GTPase activity responsible for the inactivation of KRAS, through the hydrolysis of GTP. In turn, GEFs facilitate the activation of KRAS, forcing the release of bound GDP and allowing its replacement by GTP. In mammalian cells, KRAS is normally found in its inactive state—GDP-bound [4][18][19][4,18,19].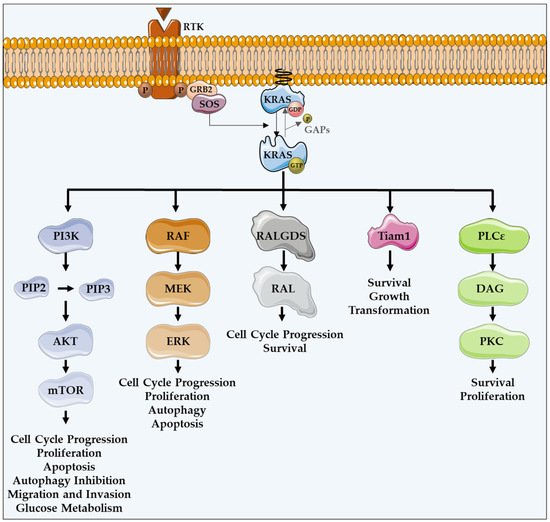
Figure 1. KRAS major effectors and cellular functions. KRAS proteins are activated upon activation of an RTK-like EGFR. KRAS is a GTPase that functions as an on/off switch that alternates between an active GTP-bound and an inactive GDP-bound state, regulated by GAPs and GEFs, such as Sos1. This oncogene regulates several effector pathways, including the MAPK and PI3K/AKT pathways. GTP-bound KRAS leads to the activation of RAF proteins, resulting in the initiation of MAPK signaling. Subsequently, MEK is activated and, in turn, phosphorylates and activates ERK. Downstream of this, ERK can regulate numerous transcription factors, promoting cell cycle progression and influencing proliferation and apoptosis. Besides MAPK pathway, KRAS can interact with PI3K, whose activation leads to the phosphorylation of PIP2, resulting in PIP3. This second messenger is able to activate a large number of proteins containing a pleckstrin homology domain, including PDK1 and AKT, whose main downstream effector is mTOR. These proteins regulate cell cycle progression, cell survival, glucose metabolism, cell growth and proliferation. In addition to MAPK and PI3K/AKT pathways, KRAS can activate RALGDS, whose downstream effector is RAL GTPases, promoting cell cycle progression and survival. Furthermore, KRAS interacts with Tiam1, a Rho family GTPase, which is implicated in the development of RAS-driven tumors, growth transformation and promotion of cell survival. KRAS also binds to PLCε 44, a phospho-lipase C isoform responsible for KRAS mediated production of DAG, resulting in calcium release and activation of the PKC signaling cascade, involved in survival, proliferation, and calcium mobilization.