 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jing-Ruey Joanna Yeh | -- | 2003 | 2022-07-14 15:58:18 | | | |
| 2 | Peter Tang | Meta information modification | 2003 | 2022-07-15 05:45:04 | | |
Video Upload Options
Aspartate is a precursor for nucleotide synthesis and is indispensable for cell proliferation. Moreover, the malate–aspartate shuttle plays a key role in redox balance, and a deficit in aspartate can lead to oxidative stress. It is now recognized that aspartate biosynthesis is largely governed by mitochondrial metabolism, including respiration and glutaminolysis in cancer cells. Therefore, under conditions that suppress mitochondrial metabolism, including mutations, hypoxia, or chemical inhibitors, aspartate can become a limiting factor for tumor growth and cancer cell survival.
1. Introduction
2. Roles of Aspartate in Cell Proliferation and Survival
2.1. Protein Synthesis, Amino Acid Metabolism, and the Urea Cycle
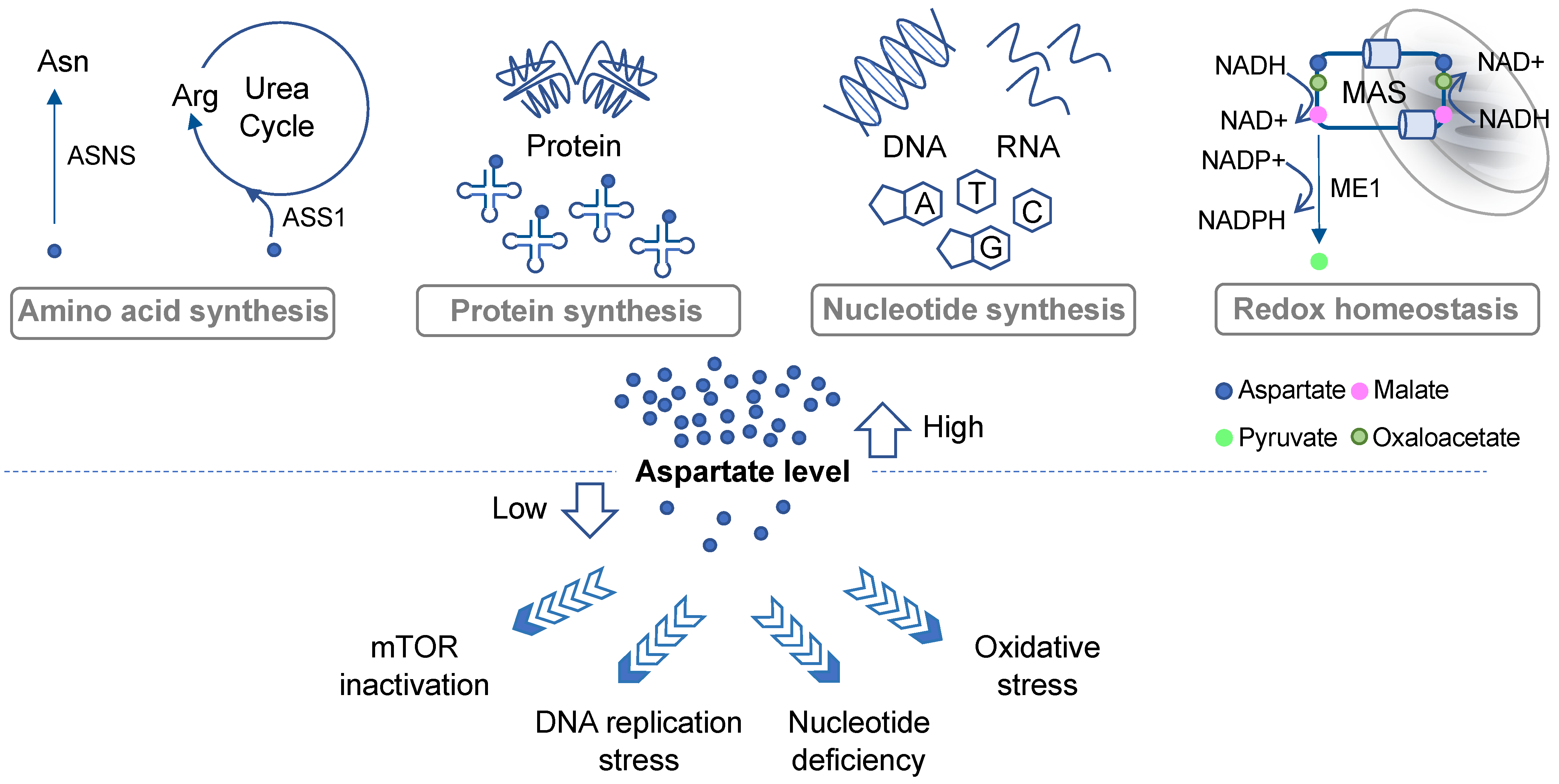
2.2. De Novo Nucleotide Synthesis
2.3. Redox Balance and Oxidative Stress
3. Aspartate Availability, Biosynthesis, and Their Limiting Steps in Cancer
3.1. Aspartate Biosynthesis in Respiration-Competent Cancers
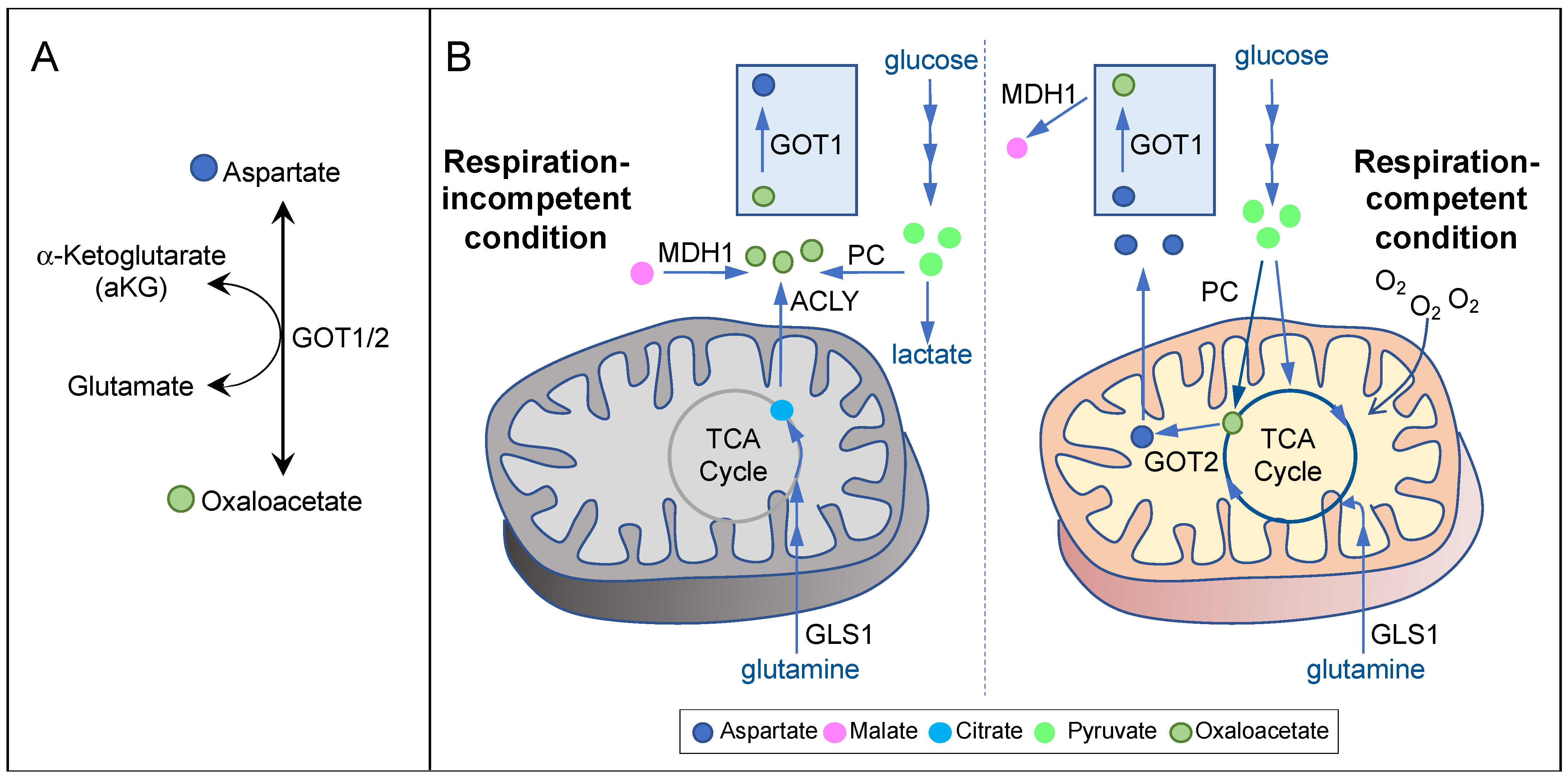
3.2. Aspartate Biosynthesis in Respiration-Incompetent Cancers
4. Therapeutic Approaches Targeting Aspartate Availability in Cancer
|
Therapeutic Agent |
Proposed Mechanism of Action |
Status of Development |
Co-Targeting Strategy |
References |
|---|---|---|---|---|
|
CB-839 |
Inhibit GLS1 |
clinical trial |
Inhibit poly(ADP-ribose) polymerase (PARP) |
[15] |
|
Metformin |
Inhibit Complex I of the electron transport chain |
FDA-approved diabetes drug |
Inhibit GOT1 or exploit GOT1 to exhaust aspartate |
|
|
Dimethyl alpha-ketoglutarate (dmaKG) |
Engage GOT1 in aspartate consumption |
Pre-clinical testing |
Inhibit mitochondrial respiration |
|
|
Chloroquine |
Unclear; possibly through inhibition of mitochondrial metabolism |
FDA-approved anti-malarial drug |
Inhibit replication stress response |
|
|
OGDH inhibition |
References
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352.
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15.
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669.
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634.
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551.
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563.
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485.
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457.
- Krall, A.S.; Mullen, P.J.; Surjono, F.; Momcilovic, M.; Schmid, E.W.; Halbrook, C.J.; Thambundit, A.; Mittelman, S.D.; Lyssiotis, C.A.; Shackelford, D.B.; et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab. 2021, 33, 1013–1026.
- Garcia-Bermudez, J.; Williams, R.T.; Guarecuco, R.; Birsoy, K. Targeting extracellular nutrient dependencies of cancer cells. Mol. Metab. 2020, 33, 67–82.
- Hettmer, S.; Schinzel, A.C.; Tchessalova, D.; Schneider, M.; Parker, C.L.; Bronson, R.T.; Richards, N.G.; Hahn, W.C.; Wagers, A.J. Functional genomic screening reveals asparagine dependence as a metabolic vulnerability in sarcoma. Elife 2015, 4, e09436.
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381.
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645.
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383.
- Okazaki, A.; Gameiro, P.A.; Christodoulou, D.; Laviollette, L.; Schneider, M.; Chaves, F.; Stemmer-Rachamimov, A.; Yazinski, S.A.; Lee, R.; Stephanopoulos, G.; et al. Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J. Clin. Investig. 2017, 127, 1631–1645.
- Madala, H.R.; Helenius, I.T.; Zhou, W.; Mills, E.; Zhang, Y.; Liu, Y.; Metelo, A.M.; Kelley, M.L.; Punganuru, S.; Kim, K.B.; et al. Nitrogen Trapping as a Therapeutic Strategy in Tumors with Mitochondrial Dysfunction. Cancer Res. 2020, 80, 3492–3506.
- Elliott, I.A.; Dann, A.M.; Xu, S.; Kim, S.S.; Abt, E.R.; Kim, W.; Poddar, S.; Moore, A.; Zhou, L.; Williams, J.L.; et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 6842–6847.
- van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J.; et al. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab. 2020, 32, 391–403.
- Taylor, E.B. Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol. 2017, 27, 633–644.
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720.
- Allen, E.L.; Ulanet, D.B.; Pirman, D.; Mahoney, C.E.; Coco, J.; Si, Y.; Chen, Y.; Huang, L.; Ren, J.; Choe, S.; et al. Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep. 2016, 17, 876–890.
- Ilic, N.; Birsoy, K.; Aguirre, A.J.; Kory, N.; Pacold, M.E.; Singh, S.; Moody, S.E.; DeAngelo, J.D.; Spardy, N.A.; Freinkman, E.; et al. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc. Natl. Acad. Sci. USA 2017, 114, E3434–E3443.
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105.
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617.
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol 2018, 20, 775–781.
- King, B.; Araki, J.; Palm, W.; Thompson, C.B. Yap/Taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes Dev. 2020, 34, 1345–1358.
- Bacci, M.; Lorito, N.; Ippolito, L.; Ramazzotti, M.; Luti, S.; Romagnoli, S.; Parri, M.; Bianchini, F.; Cappellesso, F.; Virga, F.; et al. Reprogramming of Amino Acid Transporters to Support Aspartate and Glutamate Dependency Sustains Endocrine Resistance in Breast Cancer. Cell Rep. 2019, 28, 104–118.
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350.
- Sellers, K.; Fox, M.P.; Bousamra, M., II; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698.
- Phannasil, P.; Thuwajit, C.; Warnnissorn, M.; Wallace, J.C.; MacDonald, M.J.; Jitrapakdee, S. Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells. PLoS ONE 2015, 10, e0129848.
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848.
- Phannasil, P.; Ansari, I.H.; El Azzouny, M.; Longacre, M.J.; Rattanapornsompong, K.; Burant, C.F.; MacDonald, M.J.; Jitrapakdee, S. Mass spectrometry analysis shows the biosynthetic pathways supported by pyruvate carboxylase in highly invasive breast cancer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 537–551.
- Lorendeau, D.; Rinaldi, G.; Boon, R.; Spincemaille, P.; Metzger, K.; Jager, C.; Christen, S.; Dong, X.; Kuenen, S.; Voordeckers, K.; et al. Dual loss of succinate dehydrogenase (SDH) and complex I activity is necessary to recapitulate the metabolic phenotype of SDH mutant tumors. Metab. Eng. 2016, 43, 187–197.
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 2013, 51, 236–248.
- Tyrakis, P.A.; Yurkovich, M.E.; Sciacovelli, M.; Papachristou, E.K.; Bridges, H.R.; Gaude, E.; Schreiner, A.; D’Santos, C.; Hirst, J.; Hernandez-Fernaud, J.; et al. Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep. 2017, 21, 1036–1047.
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10.
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53.
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727.
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788.
- King, M.P.; Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 1996, 264, 304–313.
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326.
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384.
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388.
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616.
- Sica, V.; Bravo-San Pedro, J.M.; Izzo, V.; Pol, J.; Pierredon, S.; Enot, D.; Durand, S.; Bossut, N.; Chery, A.; Souquere, S.; et al. Lethal Poisoning of Cancer Cells by Respiratory Chain Inhibition plus Dimethyl alpha-Ketoglutarate. Cell Rep. 2019, 27, 820–834.
- Eloranta, K.; Cairo, S.; Liljestrom, E.; Soini, T.; Kyronlahti, A.; Judde, J.G.; Wilson, D.B.; Heikinheimo, M.; Pihlajoki, M. Chloroquine Triggers Cell Death and Inhibits PARPs in Cell Models of Aggressive Hepatoblastoma. Front. Oncol. 2020, 10, 1138.


