Aspartate is a precursor for nucleotide synthesis and is indispensable for cell proliferation. Moreover, the malate–aspartate shuttle plays a key role in redox balance, and a deficit in aspartate can lead to oxidative stress. It is now recognized that aspartate biosynthesis is largely governed by mitochondrial metabolism, including respiration and glutaminolysis in cancer cells. Therefore, under conditions that suppress mitochondrial metabolism, including mutations, hypoxia, or chemical inhibitors, aspartate can become a limiting factor for tumor growth and cancer cell survival.
- oxidative phosphorylation
- mitochondrial respiration
- mitochondrial DNA mutation
- hypoxia
- aspartate
- glutaminase
- asparagine
- GOT1
- alpha-ketoglutarate
- cancer metabolism
1. Introduction
2. Roles of Aspartate in Cell Proliferation and Survival
2.1. Protein Synthesis, Amino Acid Metabolism, and the Urea Cycle
One of the important roles of aspartate in governing cell proliferation is by supporting protein and amino acid synthesis (Figure 1). Aspartate is one of the 20 proteinogenic amino acids and also serves as a precursor for the biosynthesis of asparagine and arginine. Asparate gives rise to asparagine via asparagine synthetase (ASNS). It has been shown that intracellular asparagine levels are a critical determinant of mTOR activities [16,17][8][9]. Although not all cancer cells express ASNS, such as acute lymphoblastic leukemia (ALL) blasts that mostly rely on exogenous asparagine supplies [18][10], in some cancer types ASNS expression has been linked to tumor progression [19,20][11][12]. In addition, aspartate is a nitrogen donor for arginine. Metabolism from aspartate to arginine is part of the urea cycle [21][13], whereby aspartate first reacts with citrulline via argininosuccinate synthase (ASS1) to produce argininosuccinate, which is subsequently converted into arginine and fumarate via argininosuccinate lyase (ASL). Notably, many malignancies exhibit downregulation of various urea cycle enzymes, including ASS1 [21][13]. ASS1 deficiency promotes cell proliferation by diverting aspartate for pyrimidine synthesis [22][14]. Increases in aspartate levels can also lead to activation of mTOR [22][14]. These results demonstrate that suppression of the urea cycle, even though it can cause arginine auxotrophy [21][13], provides an overall proliferation advantage to cancer cells through increased aspartate availability.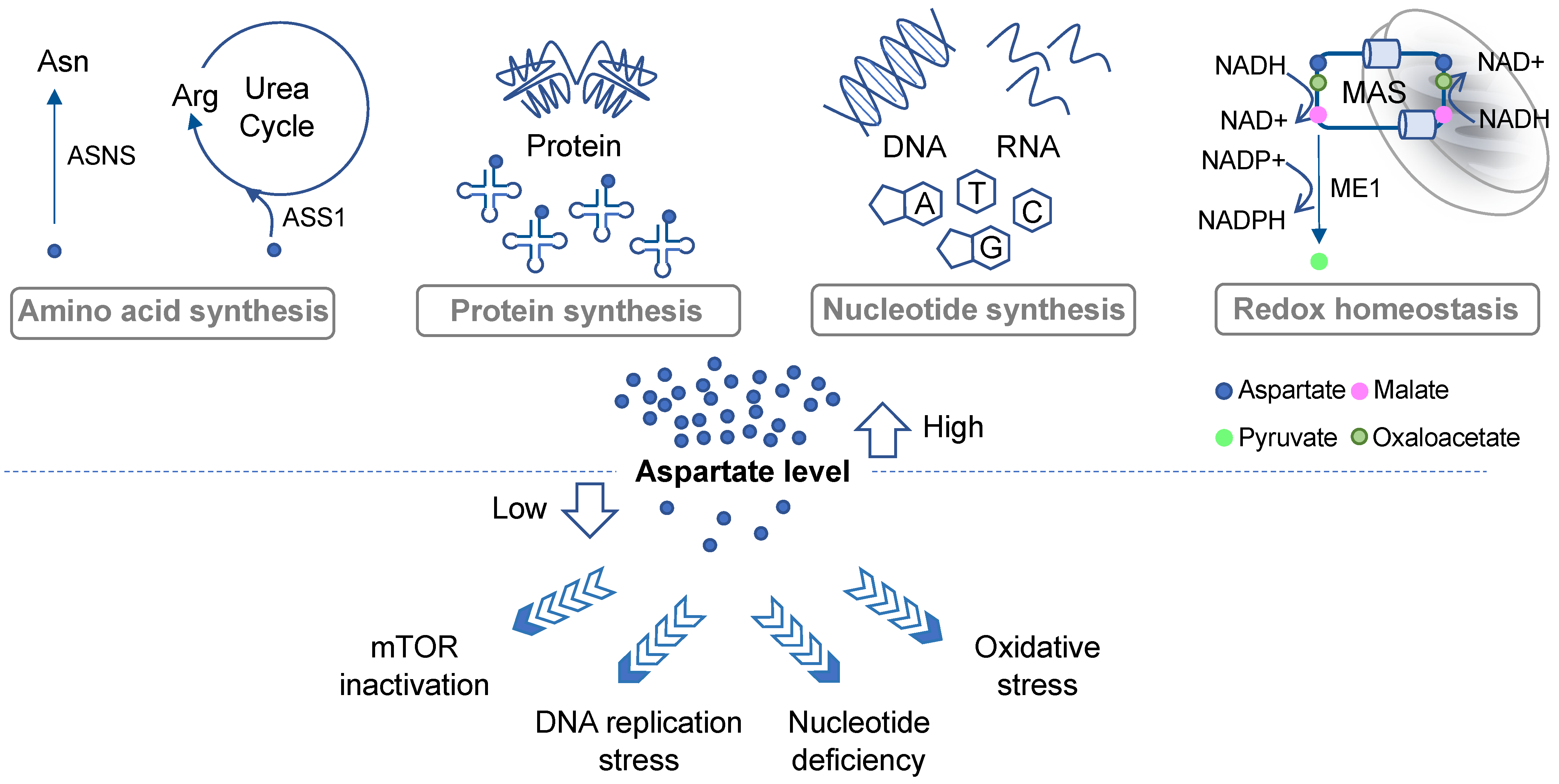
2.2. De Novo Nucleotide Synthesis
Proliferating cells have a high demand for nucleotides due to their robust DNA replication, RNA transcription, and ribosome biogenesis. Aspartate is a key building block for both purines and pyrimidines [7], contributing to three carbon atoms and one nitrogen atoms of the rings in uracil, cytosine, and thymine [7]. It also contributes to two purine biosynthesis steps including the conversions of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) into succino-AICAR (SAICAR) and of inosine monophosphate (IMP) into adenylosuccinate [7]. Aspartate deprivation can result in nucleotide deficiency, DNA replication stress, and mTOR inactivation [12,14,15][15][16][17] (Figure 1). In addition to cell proliferation, de novo nucleotide synthesis plays a critical role in cancer cells’ response to drugs. While studying the emergence of chemoresistant and relapse-initiating acute myeloid leukemia (AML) cells using a mouse model, van Gastel et al. found that induction chemotherapy could instigate significant metabolic changes in the stroma, resulting in its increased aspartate production and secretion [13][18]. Aspartate exited from the stroma could enter AML cells and increase nucleotide synthesis in these cells [13][18]. Consequently, inhibition of pyrimidine synthesis led to selective elimination of post-chemo relapse-initiating leukemia cells in the blood [13][18], suggesting that aspartate-lowering agents may in some cases reduce the advent of chemoresistance. These results further imply that investigators may need to consider the possible contribution of stroma-derived aspartate to cancer cell metabolism during the development of effective aspartate-targeting approaches.2.3. Redox Balance and Oxidative Stress
Another prominent metabolic role of aspartate is to regulate cell compartmental redox balance via the malate–aspartate shuttle (MAS) (Figure 1). The net effect of the MAS is to recycle NADH produced in the cytosol (predominantly from glycolysis) back to NAD+ and to transfer the reducing power generated in the cytosol to the mitochondrion for oxidation via oxidative phosphorylation [23][19]. The MAS utilizes two cytosolic enzymes, glutamic-oxaloacetic transaminase 1 (GOT1) and malate dehydrogenase 1 (MDH1), to convert aspartate to malate, NADH to NAD+, and alpha-ketoglutarate (aKG; also known as 2-oxoglutarate) to glutamate. It utilizes another two enzymes in the mitochondria, GOT2 and MDH2, which in a fashion that is parallel and reverse to the cytosolic reaction, convert malate to aspartate, NAD+ to NADH, and aKG to glutamate [5]. The MAS also requires two antiporters on the mitochondrial inner membrane to transport the metabolites in and out of the mitochondria [23][19]. Evidence suggests that disruption of the MAS may decrease the cytosolic NAD+/NADH ratio and increase the mitochondrial NAD+/NADH ratio, which could lead to rippling effects on other metabolic pathways [9][20]. Moreover, some cancer types, such as those carrying PI3KCA mutations, appear to be more sensitive to MAS perturbation than others [24,25][21][22]. Interestingly, in KRAS-mutant pancreatic ductal adenocarcinoma (PDAC) cells, aspartate also plays an important role in maintaining redox homeostasis by supporting NADPH production [26][23] (Figure 1). As discussed above, aspartate is an important source for malate in the cytosol. In PDAC cells, malate is utilized by NADP-dependent malic enzyme 1 (ME1) to generate pyruvate and NADPH. Inhibition of any step in the production and utilization of cytosolic malate leads to a decrease in the NADPH/NADP+ ratio, an increase in reactive oxygen species (ROS), and a significant reduction in clonogenic growth of KRAS mutant cells [26][23]. Thus, once again, this restudyearch highlights the multifaceted roles of aspartate in governing cancer cell growth and survival.3. Aspartate Availability, Biosynthesis, and Their Limiting Steps in Cancer
To target aspartate availability in cancer, it is important to know how intracellular aspartate level is supported. Aspartate concentration in human plasma is low, ranging between 0 and 68 µM [27][24]. Some cancer cells have the ability to acquire aspartate from the environment via the expression of low- or high-affinity aspartate membrane transporters, such as SLC1A2 and SLC1A3, or via macropinocytosis [8,28][25][26]. For instance, it has been shown that endocrine-resistant estrogen receptor-positive (ER+) breast cancer cells can import aspartate and glutamate, which contributes to their aggressive phenotypes and resistance to endocrine therapy [10][27]. For these cancer types, aspartate availability may be suppressed by targeting the expression or the activities of aspartate membrane transporters [10][27]. For others, it may be possible to devise an effective targeted approach by identifying the limiting steps in their respective aspartate biosynthesis pathways.3.1. Aspartate Biosynthesis in Respiration-Competent Cancers
In cells exhibiting mitochondrial respiration competency, aspartate is mainly synthesized in the mitochondria by GOT2 from glutamate and the TCA cycle metabolite oxaloacetate [5] (Figure 2A,B, right). Many cancer types use glutamine to fuel the TCA cycle [4,29][4][28]. Therefore, inhibition of glutaminase-1 (GLS1), the first step in glutaminolysis, in cancers displaying glutamine dependency may lead to aspartate deprivation. Indeed, while inhibition of GLS1 reduces aspartate production and the proliferation of von Hippel–Lindau (VHL)−/− renal cell carcinoma (RCC) cells, VHL+/+ cells can adapt by increasing glucose contribution to the TCA cycle and aspartate biosynthesis [12][15]. These results demonstrate that aspartate depletion can exert potent anticancer effects; however, it is critical that aspartate-targeting approaches should be tailored based on the specific metabolic constraints of individual cancer types.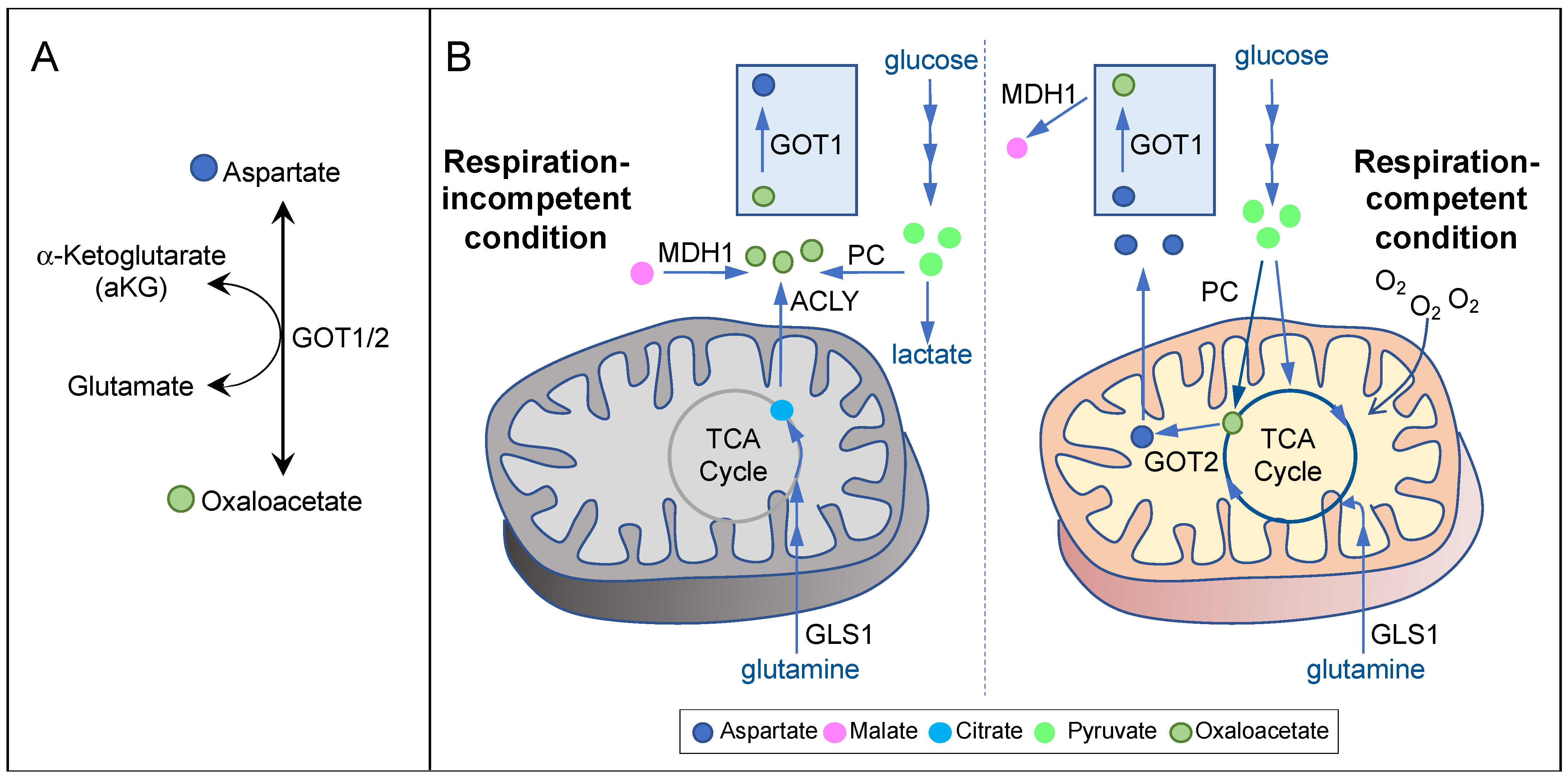
3.2. Aspartate Biosynthesis in Respiration-Incompetent Cancers
Respiration incompetency is a feature of some of the most aggressive cancer types, such as ones carrying mutations in the TCA cycle enzymes fumarate hydratase (FH) and succinate dehydrogenase (SDH) [34,35,36,37][33][34][35][36]. Tumor hypoxia or mitochondrial DNA mutations can also induce varying degrees of respiration deficiency in a wide range of cancer types [8,38][25][37]. Inhibition of mitochondrial respiration, either inducibly or inherently, can in turn cause a suppression of mitochondrial oxidative metabolism, resulting in a significant reduction in mitochondrial aspartate production [6,11][6][38]. Concordantly, aspartate is a major limiting factor for cancer cell proliferation under hypoxia or ETC inhibition both in vitro and in vivo [5,6,8,11,39][5][6][25][38][39]. Interestingly, it has been known for a long time that respiration-deficient cancer cells can proliferate in pyruvate-containing media [40]. Pyruvate can also confer resistance to OXPHOS inhibitors, implicating the presence of an alternative pathway for aspartate biosynthesis under respiration-incompetent conditions [11][38]. Indeed, Birsoy et al. showed that, in respiration-deficient cells, flux through GOT1 is reversed, so that it can generate aspartate using glutamate and oxaloacetate in the cytosol [5] (Figure 2A,B, left). In the absence of mitochondrial oxidative metabolism, oxaloacetate may be derived from three potential sources (Figure 2B, left). First, pyruvate can give rise to oxaloacetate via PC. In SDH-null cells, PC plays a pivotal role in supporting aspartate biosynthesis and cell proliferation [41]. Second, malate can give rise to oxaloacetate via MDH1 using NAD+ as the electron acceptor. Pyruvate may stimulate this reaction by recycling NADH to NAD+ via lactate dehydrogenase (LDH). In MDH1 knockout cells, pyruvate can no longer restore aspartate level in the presence of OXPHOS inhibitors [5]. Third, citrate can give rise to oxaloacetate via ATP-citrate lyase (ACLY). As citrate is generated predominantly by glutaminolysis and reductive carboxylation in respiration-impaired cells [42,43,44][42][43][44], glutamine utilization may also play a role in maintaining aspartate level in these cells. Notably, essentially all of the metabolic routes described above require GOT1 for generating aspartate under respiration incompetency. While GOT1-null cells can proliferate under respiration-competent conditions, they cannot survive OXPHOS inhibition even with pyruvate supplementation [5]. These findings strongly suggest that GOT1 may be a prime target against cancers exhibiting respiration incompetency.4. Therapeutic Approaches Targeting Aspartate Availability in Cancer
Various therapeutic approaches discussed herein are summarized in Table 1.|
Therapeutic Agent |
Proposed Mechanism of Action |
Status of Development |
Co-Targeting Strategy |
References |
|---|---|---|---|---|
|
CB-839 |
Inhibit GLS1 |
clinical trial |
Inhibit poly(ADP-ribose) polymerase (PARP) |
|
|
Metformin |
Inhibit Complex I of the electron transport chain |
FDA-approved diabetes drug |
Inhibit GOT1 or exploit GOT1 to exhaust aspartate |
|
|
Dimethyl alpha-ketoglutarate (dmaKG) |
Engage GOT1 in aspartate consumption |
Pre-clinical testing |
Inhibit mitochondrial respiration |
|
|
Chloroquine |
Unclear; possibly through inhibition of mitochondrial metabolism |
FDA-approved anti-malarial drug |
Inhibit replication stress response |
|
|
OGDH inhibition |
References
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352.
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15.
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669.
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634.
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551.
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563.
- Lane, A.N.; Fan, T.W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485.
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457.
- Krall, A.S.; Mullen, P.J.; Surjono, F.; Momcilovic, M.; Schmid, E.W.; Halbrook, C.J.; Thambundit, A.; Mittelman, S.D.; Lyssiotis, C.A.; Shackelford, D.B.; et al. Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab. 2021, 33, 1013–1026.
- Garcia-Bermudez, J.; Williams, R.T.; Guarecuco, R.; Birsoy, K. Targeting extracellular nutrient dependencies of cancer cells. Mol. Metab. 2020, 33, 67–82.
- Hettmer, S.; Schinzel, A.C.; Tchessalova, D.; Schneider, M.; Parker, C.L.; Bronson, R.T.; Richards, N.G.; Hahn, W.C.; Wagers, A.J. Functional genomic screening reveals asparagine dependence as a metabolic vulnerability in sarcoma. Elife 2015, 4, e09436.
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381.
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645.
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383.
- Okazaki, A.; Gameiro, P.A.; Christodoulou, D.; Laviollette, L.; Schneider, M.; Chaves, F.; Stemmer-Rachamimov, A.; Yazinski, S.A.; Lee, R.; Stephanopoulos, G.; et al. Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J. Clin. Investig. 2017, 127, 1631–1645.
- Madala, H.R.; Helenius, I.T.; Zhou, W.; Mills, E.; Zhang, Y.; Liu, Y.; Metelo, A.M.; Kelley, M.L.; Punganuru, S.; Kim, K.B.; et al. Nitrogen Trapping as a Therapeutic Strategy in Tumors with Mitochondrial Dysfunction. Cancer Res. 2020, 80, 3492–3506.
- Elliott, I.A.; Dann, A.M.; Xu, S.; Kim, S.S.; Abt, E.R.; Kim, W.; Poddar, S.; Moore, A.; Zhou, L.; Williams, J.L.; et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 6842–6847.
- van Gastel, N.; Spinelli, J.B.; Sharda, A.; Schajnovitz, A.; Baryawno, N.; Rhee, C.; Oki, T.; Grace, E.; Soled, H.J.; Milosevic, J.; et al. Induction of a Timed Metabolic Collapse to Overcome Cancer Chemoresistance. Cell Metab. 2020, 32, 391–403.
- Taylor, E.B. Functional Properties of the Mitochondrial Carrier System. Trends Cell Biol. 2017, 27, 633–644.
- Alkan, H.F.; Walter, K.E.; Luengo, A.; Madreiter-Sokolowski, C.T.; Stryeck, S.; Lau, A.N.; Al-Zoughbi, W.; Lewis, C.A.; Thomas, C.J.; Hoefler, G.; et al. Cytosolic Aspartate Availability Determines Cell Survival When Glutamine Is Limiting. Cell Metab. 2018, 28, 706–720.
- Allen, E.L.; Ulanet, D.B.; Pirman, D.; Mahoney, C.E.; Coco, J.; Si, Y.; Chen, Y.; Huang, L.; Ren, J.; Choe, S.; et al. Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep. 2016, 17, 876–890.
- Ilic, N.; Birsoy, K.; Aguirre, A.J.; Kory, N.; Pacold, M.E.; Singh, S.; Moody, S.E.; DeAngelo, J.D.; Spardy, N.A.; Freinkman, E.; et al. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc. Natl. Acad. Sci. USA 2017, 114, E3434–E3443.
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105.
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617.
- Garcia-Bermudez, J.; Baudrier, L.; La, K.; Zhu, X.G.; Fidelin, J.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Molina, H.; Snuderl, M.; Lewis, C.A.; et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat. Cell Biol 2018, 20, 775–781.
- King, B.; Araki, J.; Palm, W.; Thompson, C.B. Yap/Taz promote the scavenging of extracellular nutrients through macropinocytosis. Genes Dev. 2020, 34, 1345–1358.
- Bacci, M.; Lorito, N.; Ippolito, L.; Ramazzotti, M.; Luti, S.; Romagnoli, S.; Parri, M.; Bianchini, F.; Cappellesso, F.; Virga, F.; et al. Reprogramming of Amino Acid Transporters to Support Aspartate and Glutamate Dependency Sustains Endocrine Resistance in Breast Cancer. Cell Rep. 2019, 28, 104–118.
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350.
- Sellers, K.; Fox, M.P.; Bousamra, M., II; Slone, S.P.; Higashi, R.M.; Miller, D.M.; Wang, Y.; Yan, J.; Yuneva, M.O.; Deshpande, R.; et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Investig. 2015, 125, 687–698.
- Phannasil, P.; Thuwajit, C.; Warnnissorn, M.; Wallace, J.C.; MacDonald, M.J.; Jitrapakdee, S. Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells. PLoS ONE 2015, 10, e0129848.
- Christen, S.; Lorendeau, D.; Schmieder, R.; Broekaert, D.; Metzger, K.; Veys, K.; Elia, I.; Buescher, J.M.; Orth, M.F.; Davidson, S.M.; et al. Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep. 2016, 17, 837–848.
- Phannasil, P.; Ansari, I.H.; El Azzouny, M.; Longacre, M.J.; Rattanapornsompong, K.; Burant, C.F.; MacDonald, M.J.; Jitrapakdee, S. Mass spectrometry analysis shows the biosynthetic pathways supported by pyruvate carboxylase in highly invasive breast cancer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 537–551.
- Lorendeau, D.; Rinaldi, G.; Boon, R.; Spincemaille, P.; Metzger, K.; Jager, C.; Christen, S.; Dong, X.; Kuenen, S.; Voordeckers, K.; et al. Dual loss of succinate dehydrogenase (SDH) and complex I activity is necessary to recapitulate the metabolic phenotype of SDH mutant tumors. Metab. Eng. 2016, 43, 187–197.
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 2013, 51, 236–248.
- Tyrakis, P.A.; Yurkovich, M.E.; Sciacovelli, M.; Papachristou, E.K.; Bridges, H.R.; Gaude, E.; Schreiner, A.; D’Santos, C.; Hirst, J.; Hernandez-Fernaud, J.; et al. Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep. 2017, 21, 1036–1047.
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10.
- Gammage, P.A.; Frezza, C. Mitochondrial DNA: The overlooked oncogenome? BMC Biol. 2019, 17, 53.
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727.
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788.
- King, M.P.; Attardi, G. Isolation of human cell lines lacking mitochondrial DNA. Methods Enzymol. 1996, 264, 304–313.
- Cardaci, S.; Zheng, L.; MacKay, G.; van den Broek, N.J.; MacKenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326.
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384.
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388.
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616.
- Sica, V.; Bravo-San Pedro, J.M.; Izzo, V.; Pol, J.; Pierredon, S.; Enot, D.; Durand, S.; Bossut, N.; Chery, A.; Souquere, S.; et al. Lethal Poisoning of Cancer Cells by Respiratory Chain Inhibition plus Dimethyl alpha-Ketoglutarate. Cell Rep. 2019, 27, 820–834.
- Eloranta, K.; Cairo, S.; Liljestrom, E.; Soini, T.; Kyronlahti, A.; Judde, J.G.; Wilson, D.B.; Heikinheimo, M.; Pihlajoki, M. Chloroquine Triggers Cell Death and Inhibits PARPs in Cell Models of Aggressive Hepatoblastoma. Front. Oncol. 2020, 10, 1138.