Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roberta Visconti | -- | 2263 | 2022-07-13 13:26:12 | | | |
| 2 | Jessie Wu | + 1 word(s) | 2264 | 2022-07-14 03:45:28 | | | | |
| 3 | Jessie Wu | -2 word(s) | 2262 | 2022-07-15 10:31:01 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Monica, R.D.; Cuomo, M.; Buonaiuto, M.; Costabile, D.; Franca, R.A.; Caro, M.D.B.D.; Catapano, G.; Chiariotti, L.; Visconti, R. Techniques for DNA Methylation Testing in Glioblastomas. Encyclopedia. Available online: https://encyclopedia.pub/entry/25103 (accessed on 27 July 2026).
Monica RD, Cuomo M, Buonaiuto M, Costabile D, Franca RA, Caro MDBD, et al. Techniques for DNA Methylation Testing in Glioblastomas. Encyclopedia. Available at: https://encyclopedia.pub/entry/25103. Accessed July 27, 2026.
Monica, Rosa Della, Mariella Cuomo, Michela Buonaiuto, Davide Costabile, Raduan Ahmed Franca, Marialaura Del Basso De Caro, Giuseppe Catapano, Lorenzo Chiariotti, Roberta Visconti. "Techniques for DNA Methylation Testing in Glioblastomas" Encyclopedia, https://encyclopedia.pub/entry/25103 (accessed July 27, 2026).
Monica, R.D., Cuomo, M., Buonaiuto, M., Costabile, D., Franca, R.A., Caro, M.D.B.D., Catapano, G., Chiariotti, L., & Visconti, R. (2022, July 13). Techniques for DNA Methylation Testing in Glioblastomas. In Encyclopedia. https://encyclopedia.pub/entry/25103
Monica, Rosa Della, et al. "Techniques for DNA Methylation Testing in Glioblastomas." Encyclopedia. Web. 13 July, 2022.
Copy Citation
Epigenetic changes in DNA methylation contribute to the development of many diseases, including cancer. In glioblastoma multiforme, the most prevalent primary brain cancer and an incurable tumor with a median survival time of 15 months, a single epigenetic modification, the methylation of the O6-Methylguanine-DNA Methyltransferase (MGMT) gene, is a valid biomarker for predicting response to therapy with alkylating agents and also, independently, prognosis. The progress from single gene to whole-genome analysis of DNA methylation has allowed a better subclassification of glioblastomas.
glioblastoma multiforme
DNA Methylation
techniques
MGMT
whole-genome methylation profiling
1. Techniques for O6-Methylguanine-DNA Methyltransferase(MGMT) Methylation Assessment in Glioblastomas
Methylation-specific PCR (MSP) is the golden standard for MGMT methylation assessment in glioblastomas [1].
MSP can rapidly assess the methylation status of any CpG site within a CpG island [2][3]. In a first step, DNA is treated with sodium bisulfite, resulting in the conversion of unmethylated cytosine into uracil while the methylated cytosine, resistant to bisulfite, remains unaltered. Thus, MSP, as most of the techniques shown in Figure 1, utilized for investigating MGMT methylation status, requires bisulfite conversion of DNA.
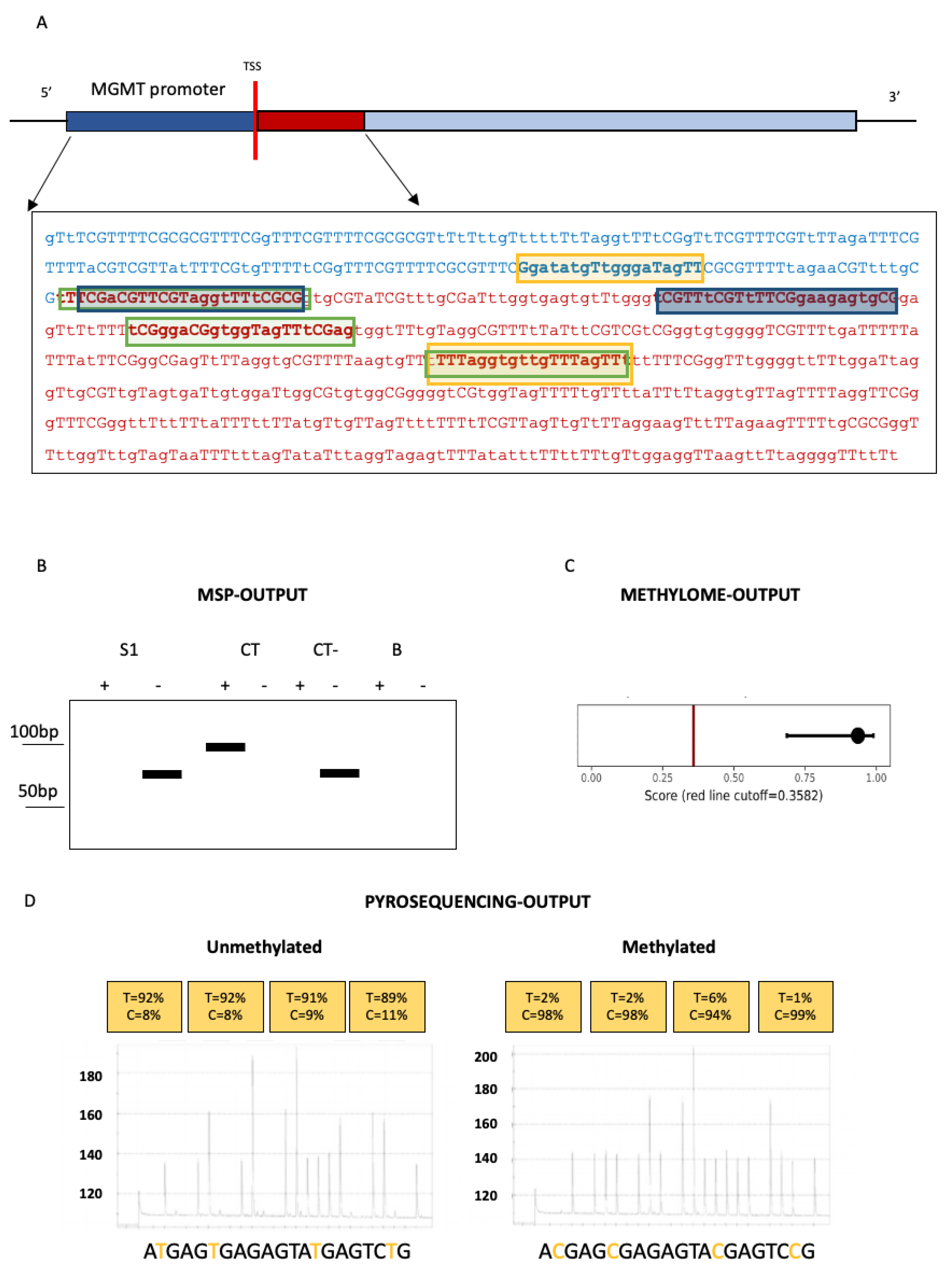
Figure 1. Graphic representation of MGMT gene regulatory region and output of the technologies used to analyze MGMT methylation after sodium bisulfite conversion. (A) MGMT gene promoter (blue) and exon 1 (red) are represented. Focus on bisulfite-converted sequence analyzed by different techniques: in yellow rectangles, primers used to perform pyrosequencing; in blue rectangles, primers used in methylation-specific PCR (MSP); in green rectangles, primers used in real-time PCR. (B) Graphical representation of MSP output: a glioblastoma sample (S1), an internal methylated (CT) and a non-methylated (CT-) controls, and a control without DNA (B) are depicted, each including a lane signed with «+» (methylated DNA specific primers) and a lane signed with «−» (un-methylated DNA specific primers). (C) Graph indicating MGMT methylation status predicted by methylome analysis. (D) Capillary electrophoresis indicating output of pyrosequencing analysis of MGMT promoter methylation in 2 glioblastoma samples.
Polymerase chain reaction amplification is then performed with two sets of primers designed to anneal to methylated cytosines or to the bisulfite-modified, unmethylated cytosines. Methylation status at a CpG site is determined by which specific primer set achieves DNA amplification. MGMT methylation status is routinely investigated in glioblastoma utilizing primers, decisively validated in clinical trials, interrogating, within exon 1, 9 CpG sites [4][5]. PCR products are, finally, run on agarose gels with appropriate negative and positive controls and samples approximately equivalent to the positive methylated control are called methylated. Thus, MSP is a qualitative technique, the results of which are based on the presence/absence of methylation in the regions where primers anneal. The technique does not allow the identification of specific methylated cytosines or even just quantification of the exact number of methylated CpG sites. Even more problematic, from a diagnostic point of view, it remains very difficult to determine validated, and standardized among different laboratories, cut-offs for calling MGMT “methylated” or “unmethylated”. However, mostly because of its simplicity and low cost, MSP is still the best method for MGMT methylation assessment in glioblastomas.
The more recently developed techniques are mostly utilized for research purposes or have been used in selected clinical trials. As an example, a real-time, methylation-specific PCR assay has been utilized in a trial designed to correlate, in newly diagnosed glioblastoma patients, the responses to dose-dense temozolomide with MGMT methylation [6]. Additionally, specificity was obtained by selective amplification of bisulfite-modified DNA, but a semiquantitative result was achieved by normalizing the number of copies of methylated MGMT to the number of copies of a housekeeping gene [7].
MGMT methylation has also been quantified through a high-resolution melt analysis, comparing differences in melting of unmethylated and methylated sequences to standards with known unmethylated to methylated ratio [8]. Although successful in predicting glioblastoma response to temozolomide, the methylation-sensitive high-resolution melt has not been validated for investigating MGMT methylation in clinical settings, thus still lacking cut-off thresholds [9].
The only quantitative technique standardized across laboratories for investigating MGMT methylation is pyrosequencing. Pyrosequencing, a “sequencing-by-synthesis” method, allows the sequencing of a single strand of DNA by synthesizing the complementary strand and detecting which base is incorporated at each step by a DNA polymerase. When used with bisulfite conversion and quantitative PCR, it can effectively determine the methylation status of each MGMT CpG dinucleotides. The concordance between MSP and pyrosequencing has been reported to be high, at least in assessing MGMT methylation in glioblastomas [10]. Accordingly, a recent prospective multicenter trial ended up showing that semiquantitative MSP and pyrosequencing are both successful in evaluating the predictive value of MGMT methylation in survival, pyrosequencing having, in addition, the greater reproducibility among the participating clinical centers. Conclusions partially disproven by three comparative studies finding that MGMT methylation status assessed by pyrosequencing is, indeed, more reliable for predicting the survival of glioblastoma patients [11][12][13]. In waiting for more conclusive studies, pyrosequencing remains, today, largely too costly to be utilized routinely in clinical settings.
All the above-mentioned techniques require DNA treatment with sodium bisulfite. In order to skip this time-consuming and often poorly efficient step, methylation-specific multiplexed ligation-dependent probe amplification (MS-MLPA) has been explored as a method for investigating MGMT methylation. MS-MLPA, the gold standard technique for studying methylation of imprinted genes in patients with suspected imprinting disorders, is based on the use of the restriction endonuclease HhaI, sensitive to methylation in its GCGC restriction site. In MS-MLPA, if the CpG locus is not methylated, the enzyme cleaves the restriction site, resulting in a lack of PCR amplification; if the CpG locus is methylated, the HhaI restriction site is not digested, resulting in the generation of a PCR product. In comparative studies, MS-MLPA has given information on MGMT methylation status concordant with that obtained with other techniques, including MSP [12][14][15][16]. Despite other noteworthy features, such as the capacity to give semiquantitative results, MS-MLPA is not, however, the standard diagnostic method for MGMT methylation status screening, this being to date, as stated above, MSP.
2. Whole-Genome Methylation Profiling (Methylome) of Glioblastomas
The whole-genome DNA methylation profile (methylome) of a tumor is the result of both somatically acquired changes and of features reflecting the cell of origin [17]. Thus, methylome analysis has been successful in both subclassifying tumors previously considered homogeneous diseases and in tracing the origin of undifferentiated metastases of cancers of unknown primary [18][19][20].
As shown in Figure 2, the entire epigenomic tumor profile can be investigated using different genome-wide, high-throughput platforms, such as the Illumina Infinium HumanMethylation450 BeadChip (450 k) or the newest Methylation BeadChip (EPIC) array, covering 850,000 CpG sites.
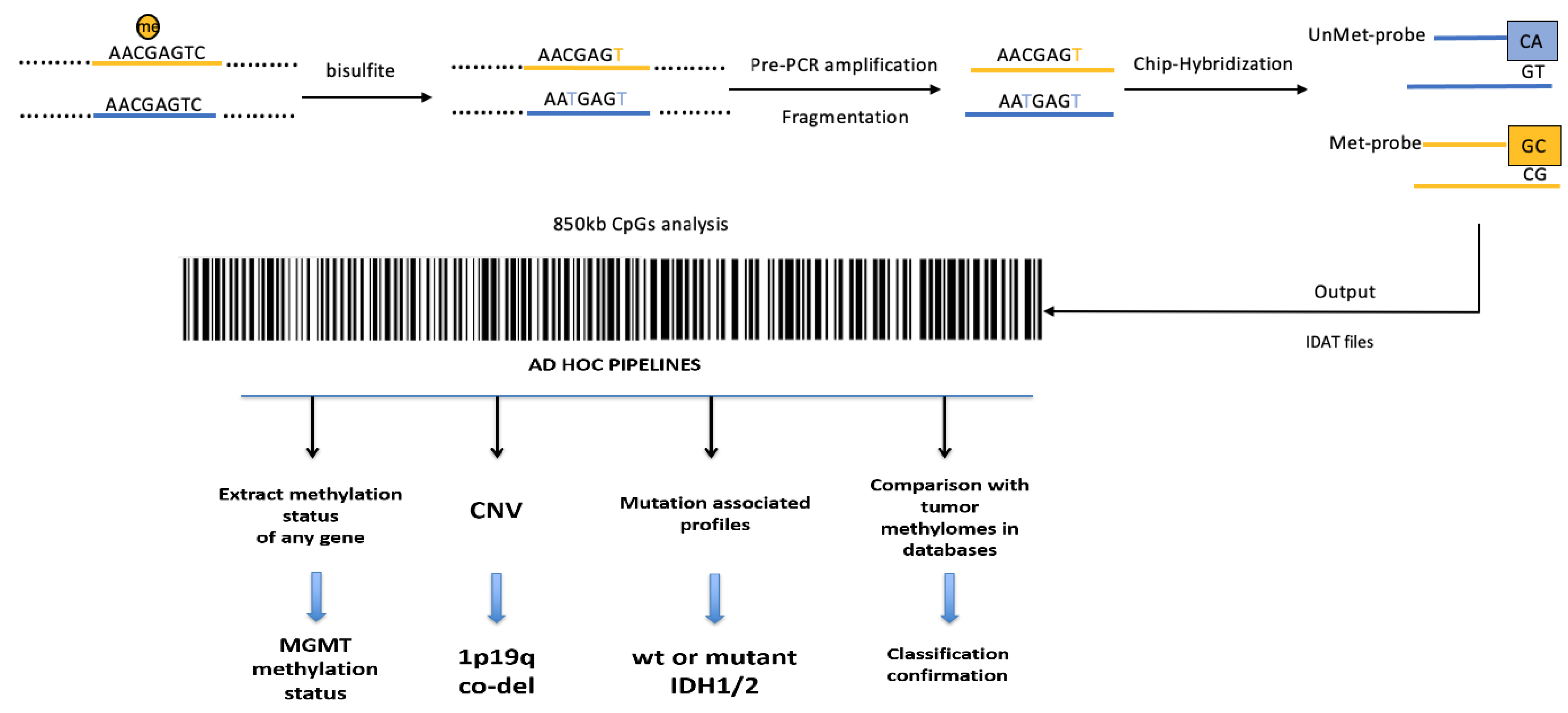
Figure 2. Methylome workflow and data analysis: schematic representation of the analytic steps to perform the EPIC array. DNA is converted with sodium bisulfite, pre-amplificated and fragmented. DNA is, then, hybridized on a specific array with specific probes recognizing the single CG both if methylated or if not methylated. The technology analyzes 850,000 CpG sites tracing a sort of tumor barcode. Output raw data are analyzed using different bioinformatic pipelines to extrapolate information useful for the characterization of the tumor, such as the methylation status of the MGMT gene, the copy number variations (CNVs), the co-deletion 1p-19q, the presence of functional mutations in the Isocitrate Dehydrogenase (IDH) 1 and 2 genes. Using a specific comparison algorithm, the analyzed methylomic profile is compared with others present in the database for a more precise classification of the tumor.
In a pivotal paper, the re-classification of CNS tumors on the basis of their methylation signature resulted in a change of diagnosis in up to 12% of the cases, demonstrating how informative the methylome is [21]. Moreover, using ad hoc bioinformatic pipelines, information can be extrapolated, by the whole-genome data, about the methylation status of every single gene, including MGMT, and about gene “copy number variation” (CNV). By CNV analysis, large chromosomal rearrangements and loss or acquisition of material for single genes and/or entire chromosomes can be readily recognized. More narrowing bioinformatic pipelines can be used to also assess the co-deletion 1p-19q, a prerequisite for oligodendroglioma diagnosis, or the therapeutically targetable Epidermal Growth Factor Receptor (EGFR) amplifications.
Methylome profiling of glioblastoma is costly and technically challenging, and its reliability also depends on the percentage of tumor cells present in the analyzed tissue. On the one hand, thus, the quantitative measurement of DNA methylation by genome-wide, high-throughput platforms is not, at the moment, affordable for all the clinical centers; on the other hand, it is a plus that some centers, including ours, do use in clinical practice to give more, useful information for glioblastoma management [1][22].
To improve the whole-genome DNA methylation-based classification of glioblastomas, the pitfall of their highly heterogenicity has to be further dealt with. In glioblastoma, tumor cells with different characteristics have been identified, recapitulating neural development and, thus, named, on the basis of their major genotypic and phenotypic features, as neural-progenitor-like, oligodendrocyte-progenitor-like, astrocyte-like and mesenchymal-like cells [23][24]. Each glioblastoma comprises cells belonging to the four types, although at different frequencies. Adding complication, during tumorigenesis, plasticity has been demonstrated between tumor cell types, modulated by genetic drivers and influenced by the tumor microenvironment [24]. Thus, glioblastomas are highly heterogenous, both at the molecular and at the cellular level, and there is high variability both within and between tumors.
Additionally, DNA methylation is highly variable in glioblastomas, with, remarkably, some samples exhibiting higher differences in DNA methylation within tumors than between tumors. Moreover, within a tumor, a high percentage of CpG sites have different methylation levels [25]. Such a high variability has to be borne in mind when DNA methylation is used for tumor classification and subtyping. Numerous approaches are under investigation to tackle the problem, including, intuitively, the analysis of more than one biopsy taken from different areas within the tumor mass or the more complex studies on single tumor cells, expanded in culture as single-cell clones [26][27]. Still, to date, glioblastoma intratumor heterogeneity negatively impacts the methylome profile-based classification, and more research is needed to further improve the usefulness of these molecular investigations for clinicians. Moreover, whole-genome analyses also need improvement to address the role of non-CpG methylation in the development and progression of cancer, including glioblastoma [28]. Parenthetically, glioblastoma heterogenicity can also affect the prognostic/predictive value of the MGMT gene methylation status [29]. To improve, a novel analysis, considering separate cut-off values for calling each individual CpG as methylated or unmethylated in the MGMT gene, has been proposed [30][31].
3. DNA Methylation Analysis of Glioblastomas by Nanopore, “Third-Generation” Sequencing
Nanopore sequencing, as shown in Figure 3, is a new technology that works by registering changes to an electrical current as nucleic acids, DNA or RNA pass through a protein nanopore [32]. The resulting signal is directly decoded to provide the specific sequence.
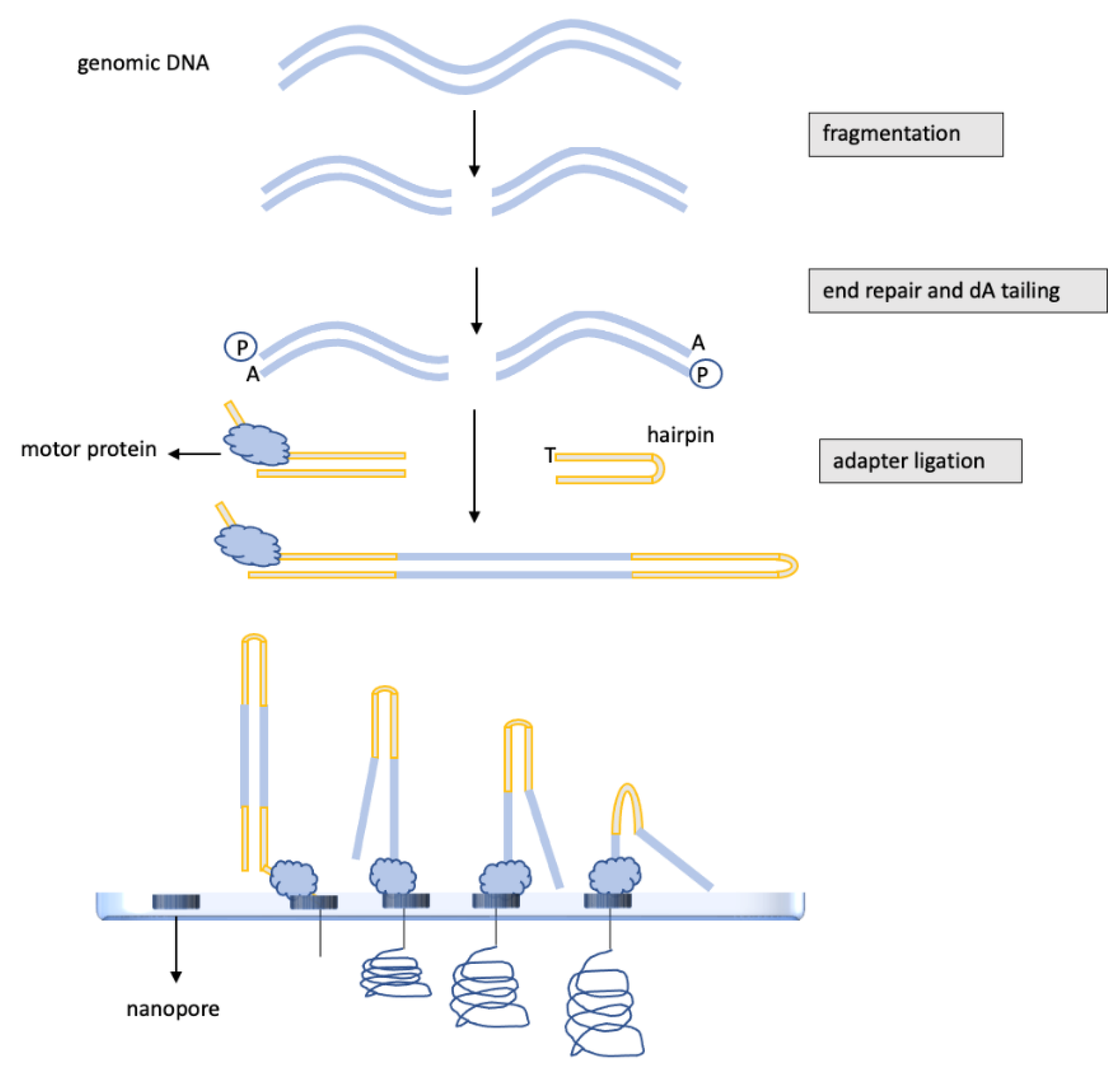
Figure 3. Nanopore sequencing strategy. As the first step, the genomic DNA is fragmented and then subjected to end repair and dA tailing. To allow DNA passage through flow cell pores, DNA is ligated to a protein adapter. Each flow cell may contain from 512 to 10,700 channels that may be potentially suitable for sequencing from 10–20 Gigabases to up to 100 Gigabases. Each channel is linked to a specific electrode, and a constant voltage is applied across the membrane. When double-stranded DNA molecules cross the pores, the voltage changes according to the specific single nucleotide passing the membrane. The difference in electric potential between the two sides of the membrane is converted into the specific nucleotide that may be identified in real-time during nanopore runs.
Thus, nanopore sequencers allow real-time analysis of DNA or RNA fragments and, importantly, provide the longest read lengths, up to 2 Mb. Moreover, the library preparation is relatively easy and of great relevance for DNA methylation studies; nanopore sequencers can detect 5-methylcytosine modifications in native DNA without the need for the time-consuming and often inefficient bisulfite conversion. Even though the smaller, more handy devices, such as the portable MinION, yield low-coverage sequences, first evidence has been published showing how nanopore technology, if well implemented, can be utilized for brain tumor characterization. A pilot study on 45 glioma samples has demonstrated that nanopore sequencing allowed to classify brain tumors on the basis of the whole-genome DNA methylation profile with precision comparable to the EPIC array [33]. Moreover, nanopore detected MGMT promoter methylation with the same accuracy as pyrosequencing and EPIC array in all the investigated cases [33]. Thus, in the near future, nanopore sequencing might indeed be a lower cost and less time-consuming alternative method for MGMT gene methylation assessment and for methylome-based classification of glioblastomas.
Nanopore potentials have been further improved by the nanopore Cas9-targeted sequencing (nCATS) strategy. nCATS uses Cas9 to specifically target and cut chromosomal DNA, then ligate adapters for nanopore sequencing. nCATS can simultaneously assess single-nucleotide variants, structural variations and CpG methylation [34]. Accordingly, nCATS has been successful in simultaneously detecting methylation of the MGMT gene and mutations in Isocitrate Dehydrogenase (IDH) 1/2 genes, these necessary for better narrowing glioblastoma diagnosis [35].
The nanopore’s peculiar ability to generate sequences in real-time and, moreover, without the need of the time-consuming bisulfite DNA conversion procedures, opens even more opportunities for better managing brain tumors. Thus, nanopore has been successfully used to obtain intraoperatively whole-genome methylation profiles of brain tumors, including glioblastomas [36]. Intraoperative nanopore sequencing combined with machine learning diagnostics has allowed tumor classification, concordant with those obtained upon a complete, standard neuropathological evaluation, in 89% of the cases [36]. Importantly, the results were returned to the neurosurgeon at a median of 97 min [36]. Knowing intraoperatively precise tumor typing is key for the surgeon to decide the better surgical strategy, a great help in choosing between maximal resection, whose benefits depend, indeed, on the tumor characteristic, and the risk of severe brain damage. Strikingly, an intraoperative diagnosis of a low-grade glial-neuronal tumor can even lead to the decision that cytoreduction is not indicated. On the other hand, multiple lesions, intraoperatively diagnosed as multifocal diffuse gliomas, can stop the surgeon, as the risks of radical resections outweigh the expected benefits.
Besides the more futuristic applications such as the intraoperative use, overall, it is predictable that nanopore sequencing, thanks to its peculiar ability to provide, more quickly and effortlessly, information on gene mutation and methylation, will be soon a broadly used alternative to more conventional sequencing methods.
References
- Philteos, J.; Karmur, B.S.; Mansouri, A. MGMT testing in glioblastomas: Pitfalls and opportunities. Am. J. Clin. Oncol. 2019, 42, 117–122.
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826.
- Della Monica, R.; Cuomo, M.; Visconti, R.; di Mauro, A.; Buonaiuto, M.; Costabile, D.; De Riso, G.; Di Risi, T.; Guadagno, E.; Tafuto, R.; et al. Evaluation of MGMT gene methylation in neuroendocrine neoplasms. Oncol. Res. 2022, 28, 837–845.
- Hegi, M.E.; Diserens, A.C.; Godard, S.; Dietrich, P.Y.; Regli, L.; Ostermann, S.; Otten, P.; Van Melle, G.; de Tribolet, N.; Stupp, R. Clinical trial substantiates the predictive value of 0-6-methylguanine-DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin. Cancer Res. 2004, 10, 871–874.
- Esteller, M.; Hamilton, S.R.; Burger, P.C.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999, 59, 793–797.
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-dense temozolomide for newly diagnosed glioblastoma: A randomized phase III clinical trial. J. Clin. Oncol. 2013, 31, 4085–4091.
- Vlassenbroeck, I.; Califice, S.; Diserens, A.C.; Migliavacca, E.; Straub, J.; Di Stefano, I.; Moreau, F.; Hamou, M.F.; Renard, I.; Delorenzi, M.; et al. Validation of real-time methylation-specific PCR to determine O6-methylguanine-DNA methyltransferase gene promoter methylation in glioma. J. Mol. Diagn. 2008, 10, 332–337.
- Wojdacz, T.K.; Dobrovic, A. Methylation-sensitive high resolution melting (MS-HRM): A new approach for sensitive and high-throughput assessment of methylation. Nucleic Acids Res. 2007, 35, e41.
- Switzeny, O.J.; Christmann, M.; Renovanz, M.; Giese, A.; Sommer, C.; Kaina, B. MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clin. Epigenetics 2016, 8, 49.
- Estival, A.; Sanz, C.; Ramirez, J.L.; Velarde, J.M.; Domenech, M.; Carrato, C.; de Las Peñas, R.; Gil-Gil, M.; Sepúlveda, J.; Armengol, R.; et al. Pyrosequencing versus methylation-specific PCR for assessment of MGMT methylation in tumor and blood samples of glioblastoma patients. Sci. Rep. 2019, 9, 11125.
- Karayan-Tapon, L.; Quillien, V.; Guilhot, J.; Wager, M.; Fromont, G.; Saikali, S.; Etcheverry, A.; Hamlat, A.; Loussouarn, D.; Campion, L.; et al. Prognostic value of O6-methylguanine-DNA methyltransferase status in glioblastoma patients, assessed by five different methods. J. Neuro-Oncology 2010, 97, 311–322.
- Christians, A.; Hartmann, C.; Benner, A.; Meyer, J.; von Deimling, A.; Weller, M.; Wick, W.; Weiler, M. Prognostic value of three different methods of MGMT promoter methylation analysis in a prospective trial on newly diagnosed glioblastoma. PLoS ONE 2012, 7, e33449.
- Quillien, V.; Lavenu, A.; Karayan-Tapon, L.; Carpentier, C.; Labussière, M.; Lesimple, T.; Chinot, O.; Wager, M.; Honnorat, J.; Saikali, S.; et al. Comparative assessment of 5 methods (methylation-specific polymerase chain reaction, MethyLight, pyrosequencing, methylation-sensitive high-resolution melting, and immunohistochemistry) to analyze O6-methylguanine-DNA-methyltranferase in a series of 100 glioblastoma patients. Cancer 2012, 118, 4201–4211.
- Jeuken, J.W.; Cornelissen, S.J.; Vriezen, M.; Dekkers, M.M.; Errami, A.; Sijben, A.; Boots-Sprenger, S.H.; Wesseling, P. MS-MLPA: An attractive alternative laboratory assay for robust, reliable, and semiquantitative detection of MGMT promoter hypermethylation in gliomas. Lab. Investig. 2007, 87, 1055–1065.
- Shah, N.; Lin, B.; Sibenaller, Z.; Ryken, T.; Lee, H.; Yoon, J.G.; Rostad, S.; Foltz, G. Comprehensive analysis of MGMT promoter methylation: Correlation with MGMT expression and clinical response in GBM. PLoS ONE 2011, 6, e16146.
- Trabelsi, S.; Mama, N.; Ladib, M.; Karmeni, N.; Haddaji Mastouri, M.; Chourabi, M.; Mokni, M.; Tlili, K.; Krifa, H.; Yacoubi, M.T.; et al. MGMT methylation assessment in glioblastoma: MS-MLPA versus human methylation 450K beadchip array and immunohistochemistry. Clin. Transl. Oncol. 2016, 18, 391–397.
- Mancarella, D.; Plass, C. Epigenetic signatures in cancer: Proper controls, current challenges and the potential for clinical translation. Genome Med. 2021, 13, 23.
- Orjuela, S.; Menigatti, M.; Schraml, P.; Kambakamba, P.; Robinson, M.D.; Marra, G. The DNA hypermethylation phenotype of colorectal cancer liver metastases resembles that of the primary colorectal cancers. BMC Cancer 2020, 20, 290.
- Modhukur, V.; Sharma, S.; Mondal, M.; Lawarde, A.; Kask, K.; Sharma, R.; Salumets, A. Machine learning approaches to classify primary and metastatic cancers using tissue of origin-based DNA methylation profiles. Cancers 2021, 13, 3768.
- Smit, K.N.; Boers, R.; Vaarwater, J.; Boers, J.; Brands, T.; Mensink, H.; Verdijk, R.M.; van Ijcken, W.F.J.; Gribnau, J.; de Klein, A.; et al. Genome-wide aberrant methylation in primary metastatic UM and their matched metastases. Sci. Rep. 2022, 12, 42.
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. A methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474.
- Epigenetic Lab—CEINGE, Advanced Biotechnologies. Available online: https://www.ceinge.unina.it/en/epigenetic-lab (accessed on 9 May 2022).
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401.
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849.
- Wenger, A.; Ferreyra Vega, S.; Kling, T.; Bontell, T.O.; Jakola, A.S.; Carén, H. Intratumor DNA methylation heterogeneity in glioblastoma: Implications for DNA methylation-based classification. Neuro-Oncology 2019, 21, 616–627.
- Parker, N.R.; Hudson, A.L.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Cheng, Z.J.; Vafaee, F.; Chen, J.; et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci. Rep. 2016, 6, 22477.
- Akgül, S.; Patch, A.M.; D’Souza, R.C.J.; Mukhopadhyay, P.; Nones, K.; Kempe, S.; Kazakoff, S.H.; Jeffree, R.L.; Stringer, B.W.; Pearson, J.V.; et al. Intratumoural heterogeneity underlies distinct therapy responses and treatment resistance in glioblastoma. Cancers 2019, 11, 190.
- Ramasamy, D.; Deva Magendhra Rao, A.K.; Rajkumar, T.; Mani, S. Non-CpG methylation—A key epigenetic modification in cancer. Brief. Funct. Genom. 2021, 20, 304–311.
- Brigliadori, G.; Goffredo, G.; Bartolini, D.; Tosatto, L.; Gurrieri, L.; Mercatali, L.; Ibrahim, T. Influence of intratumor heterogeneity on the predictivity of MGMT gene promoter methylation status in glioblastoma. Front. Oncol. 2020, 10, 533000.
- Chai, R.-C.; Liu, Y.-Q.; Zhang, K.-N.; Wu, F.; Zhao, Z.; Wang, K.-Y.; Jiang, T.; Wang, Y.-Z. A novel analytical model of MGMT methylation pyrosequencing offers improved predictive performance in patients with gliomas. Mod. Pathol. 2019, 32, 4–15.
- Pezone, A.; Tramontano, A.; Scala, G.; Cuomo, M.; Riccio, P.; De Nicola, S.; Porcellini, A.; Chiariotti, L.; Avvedimento, E.V. Tracing and tracking epiallele families in complex DNA populations. NAR Genom. Bioinform. 2020, 2, lqaa096.
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30.
- Patel, A.; Dogan, H.; Payne, A.; Krause, E.; Sievers, P.; Schoebe, N.; Schrimpf, D.; Blume, C.; Stichel, D.; Holmes, N.; et al. Rapid-CNS2: Rapid comprehensive adaptive nanopore-sequencing of CNS tumors, a proof-of-concept study. Acta Neuropathol. 2022, 143, 609–612.
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438.
- Wongsurawat, T.; Jenjaroenpun, P.; De Loose, A.; Alkam, D.; Ussery, D.W.; Nookaew, I.; Leung, Y.K.; Ho, S.M.; Day, J.D.; Rodriguez, A. A novel Cas9-targeted long-read assay for simultaneous detection of IDH1/2 mutations and clinically relevant MGMT methylation in fresh biopsies of diffuse glioma. Acta Neuropathol. Commun. 2020, 8, 87.
- Djirackor, L.; Halldorsson, S.; Niehusmann, P.; Leske, H.; Capper, D.; Kuschel, L.P.; Pahnke, J.; Due-Tønnessen, B.J.; Langmoen, I.A.; Sandberg, C.J.; et al. Intraoperative DNA methylation classification of brain tumors impacts neurosurgical strategy. Neuro-Oncology Adv. 2021, 3, vdab149.
More
Information
Subjects:
Oncology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
3 times
(View History)
Update Date:
15 Jul 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No