Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bi-He Cai | -- | 1409 | 2022-07-07 12:20:43 | | | |
| 2 | Peter Tang | Meta information modification | 1409 | 2022-07-08 02:39:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cai, B.; Hsu, Y.; Yeh, F.; Lin, Y.; Lu, R.; Yu, S.; Shaw, J.; Wu, M.; Tsai, Y.; Lin, Y.; et al. p53 Mutations in Cancers. Encyclopedia. Available online: https://encyclopedia.pub/entry/24905 (accessed on 23 July 2026).
Cai B, Hsu Y, Yeh F, Lin Y, Lu R, Yu S, et al. p53 Mutations in Cancers. Encyclopedia. Available at: https://encyclopedia.pub/entry/24905. Accessed July 23, 2026.
Cai, Bi-He, Yun-Chien Hsu, Fang-Yu Yeh, Yu-Rou Lin, Rui-Yu Lu, Si-Jie Yu, Jei-Fu Shaw, Ming-Han Wu, Yi-Zhen Tsai, Ying-Chen Lin, et al. "p53 Mutations in Cancers" Encyclopedia, https://encyclopedia.pub/entry/24905 (accessed July 23, 2026).
Cai, B., Hsu, Y., Yeh, F., Lin, Y., Lu, R., Yu, S., Shaw, J., Wu, M., Tsai, Y., Lin, Y., Bai, Z., Shih, Y., Hsu, Y., Liao, R., Kuo, W., Hsu, C., Lien, C., & Chen, C. (2022, July 07). p53 Mutations in Cancers. In Encyclopedia. https://encyclopedia.pub/entry/24905
Cai, Bi-He, et al. "p53 Mutations in Cancers." Encyclopedia. Web. 07 July, 2022.
Copy Citation
The members of the p53 family comprise p53, p63, and p73, and full-length isoforms of the p53 family have a tumor suppressor function. However, p53, but not p63 or p73, has a high mutation rate in cancers causing it to lose its tumor suppressor function.
p53
mutation
gain of function
anti-cancer drugs
1. Introduction to the p53 Family
The p53 family has three members, p53, p63, and p73 [1][2][3]. TA (transactivation) isoforms of p53 family members are tumor suppressor genes [4][5]. p53 has a high frequency of mutation in cancers causing loss of its tumor suppression function [6][7]; however, p63 and p73 are rarely mutated in cancers [8][9][10].
1.1. p53
p53 was the second tumor suppressor gene identified, although p53 was actually discovered in 1979, before the first tumor suppressor gene Rb, which was cloned in 1986 [11]. p53 has a function in apoptosis, cell cycle arrest, autophagy, metabolism, DNA repair, translational control, and feedback mechanisms [12][13]. p53 knockout mice are prone to the spontaneous variety of tumors by 6 months of age (~age 34 in humans) [14]. p53 has an average ~50% mutation rate in cancers [6][7][15]. According to the Catalogue of Somatic Mutations in Cancer (COSMIC) database [16], there are 177,561 unique clinical samples with 50,215 unique samples having p53 mutations (Figure 1A). The top p53 mutation type is missense mutation accounting for 62.74% of mutations, and the second most prevalent mutation of p53 is a nonsense mutation which accounts for 10.72% of mutations. Eight missense mutations R175H, G245S, R248Q, R248W, R249S, R273H, R273S, and R282W called hotspot mutations account for ~28% of all p53 mutations identified in cancers [17]. p53 germline mutation can cause Li-Fraumeni syndrome (LFS) which is a hereditary syndrome with a relatively early age of cancer diagnosis usually before the age of 36 [18]. This syndrome is characterized by the early onset of various types of cancer such as soft-tissue, breast, brain, leukemia, lymphoma, gastrointestinal, head and neck, kidney, larynx, lung, skin, ovary, pancreas, prostate, testis, thyroid, and adrenocortical cancers [19][20]. Some congenital p53 mutations of the LFS are similar to those acquired by p53 hotspot mutations in cancers such as G245S [21][22], but some congenital p53 mutations only appear in LFS as germ-line specific mutations such as R337H [23][24].
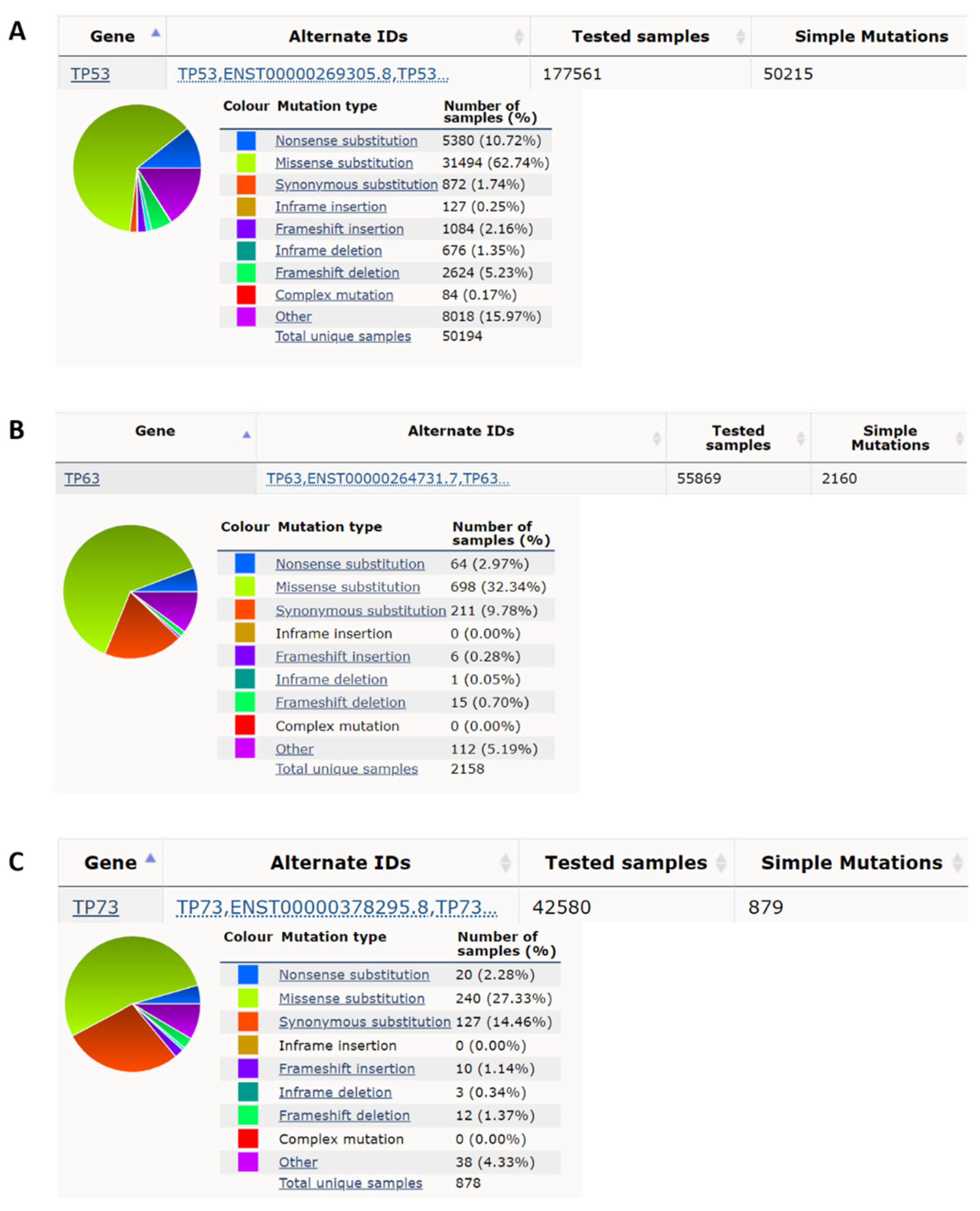
Figure 1. p53 has a high nonsense and missense mutation rate compared to p63 and p73 in cancer. (A) p53 has, respectively, a 10.72% and 62.74% nonsense mutation rate and missense mutation rate in all cancer mutation samples from the COSMIC database (https://cancer.sanger.ac.uk/cosmic; accessed date (29 April 2022) [16]. (B) p63 has, respectively, 2.97% and 32.34% nonsense mutation rate and missense mutation rate in all cancer mutation samples. (C) p73 has, respectively, 2.28% and 27.33%, nonsense mutation rate and missense mutation rate in all cancer mutation samples.
2. Types of p53 Mutations in Cancers
2.1. Nonsense Mutation
p53 nonsense mutations comprise ~10% of all p53 mutants (Figure 1A); the actual p53 nonsense mutation rate is higher than the average ~5% [25]. There are three pre-stop DNA codons, TAA, TAG, and TGA. Nonsense mutation leads to the generation of premature termination codons (PTC), which leads to nonsense-mediated mRNA decay (NMD), resulting in the inability to express full-length proteins and extremely low expression levels of truncated proteins [26]. Two mechanisms are known to regenerate full-length proteins, one is to inhibit NMD, and the other is for PTC readthrough. It is known that aminoglycoside drugs, such as G418 and gentamicin, can inhibit NMD and promote p53 PTC readthrough [27][28]. But these drugs are highly toxic and cannot be used in clinical practice. 2,6-Diaminopurine (DAP), can inhibit the activity of putative ribosomal RNA methyltransferase 1 (FTSJ1) to increase the capacity of tRNATrp to recognize the UGA stop codon to promote p53 PTC readthrough, but this drug does not have the ability to inhibit NMD [29]. Furthermore, DAP is only effective for nonsense mutations of TGA but not TAA or TAG [29], and this greatly reduces the available targets. In addition, some phthalimide derivatives and antimalarial drug quinines can promote the p53 PTC read-through ability of G418 to increase the proportion of full-length p53 and to reduce the expression of the truncated protein, but these drugs alone have no effect on PTC read-through [30][31]. Non-aminoglycoside drugs, such as Ataluren (PTC124), also increase the read-through ability of PTC without the ability to inhibit NMD, and PTC124 has been used in clinical phase II or III trials to treat genetic diseases with specific nonsense mutations [32][33]. PTC124 can also promote p53 PTC readthrough [34]. A recent study has shown that CC-885 and CC-90009 can inhibit NMD; of note, the effective concentration of CC-885 for treatment of p53 nonsense mutation with TAA is only one-tenth of that of CC-90009 [35].
2.2. Loss-of-Function Mutants
p53 missense mutations contain both loss of function and gain of function. p53 mutation is mainly located at the N-terminal transactivation domain or middle DNA binding domain [2]. In addition, several point mutants still have normal DNA binding function [36][37][38][39]; most p53 mutations within the N-terminal transactivation domain or the DNA binding domain lose their transactivation function or DNA binding function causing loss of their tumor suppressor functions such as cell cycle checkpoint controls and apoptosis [40][41][42]. These p53 mutations can associate with p63 and p73, whereas wild-type p53 cannot [43][44]. Therefore, loss-of-function p53 mutants act in the same way as the ∆N isofroms of the p53 family having a dominant-negative effect to repress the functions of normal TA isoforms of p53 family members [44][45].
2.3. Gain-of-Function Mutations
Some p53 mutants can obtain some oncogenic functions such as cell migration, invasion, and metastasis to enhance tumorigenesis [46], and these p53 mutants are called gain-of-function mutants. The acquisition of p53 gain-of-function is via three mechanisms [47][48]. First, mutant p53 can directly bind to the novel binding site with a p53 non-canonical sequence to activate several oncogenic genes [38]. Second, mutant p53 can act as a co-activator to bind to other transcription factors to activate some oncogenic genes [49]. Third, mutant p53 can bind to other tumor suppressive-type transcription factors to cause loss of transcription ability [50]. Some p53 mutants can become aggregated in several types of cancer, such as breast, lung ovary, colorectal, and head and neck cancers [51][52][53][54]. It is known that these aggregations of mutant p53 can sequester other tumor suppressor genes as a third mechanism to cause p53 gain of function [55][56][57].
3. Factors Influencing p53 Mutant Gain of Function
Several factors have been reported to influence p53 mutant gain of function (Figure 2A). The heat shock protein 70 (HSP70) has been reported to enhance mutant p53 aggregation [58], and heat shock protein 90 (HSP90) can repress mutant p53 aggregation [58][59]. SIRT1 is an NAD+ dependent histone deacetylase that has been reported to deacetylate HSF1 to enhance HSF1 transcriptional activity to increase HSP70 expression [60]. NAMPT can enhance SIRT1 activity by increasing the amount of NAD+ [61]. p53 gain-of-function mutant can induce MYC [49][62], and MYC can enhance NAPMT [63]. Wild-type p53 can induce 14-3-3σ expression [64][65], and 14-3-3σ can promote MYC poly-ubiquitination and degradation [66]. Another article also reported that wild-type p53 can bind to G-quadruplexes on MYC promoter to repress MYC expression [67]. Both wild-type and non-gain-of-function p53 mutants can activate lincRNA-p21 through binding to G-quadruplexes on lincRNA-p21 promoter [68], and lincRNA-p21 can repress STAT3 [69]. STAT3 can bind to the HSP70 promoter to active HSP70 expression [70][71]. The relationships between factors that influence p53 mutant gain of function are summarized in Figure 2A.
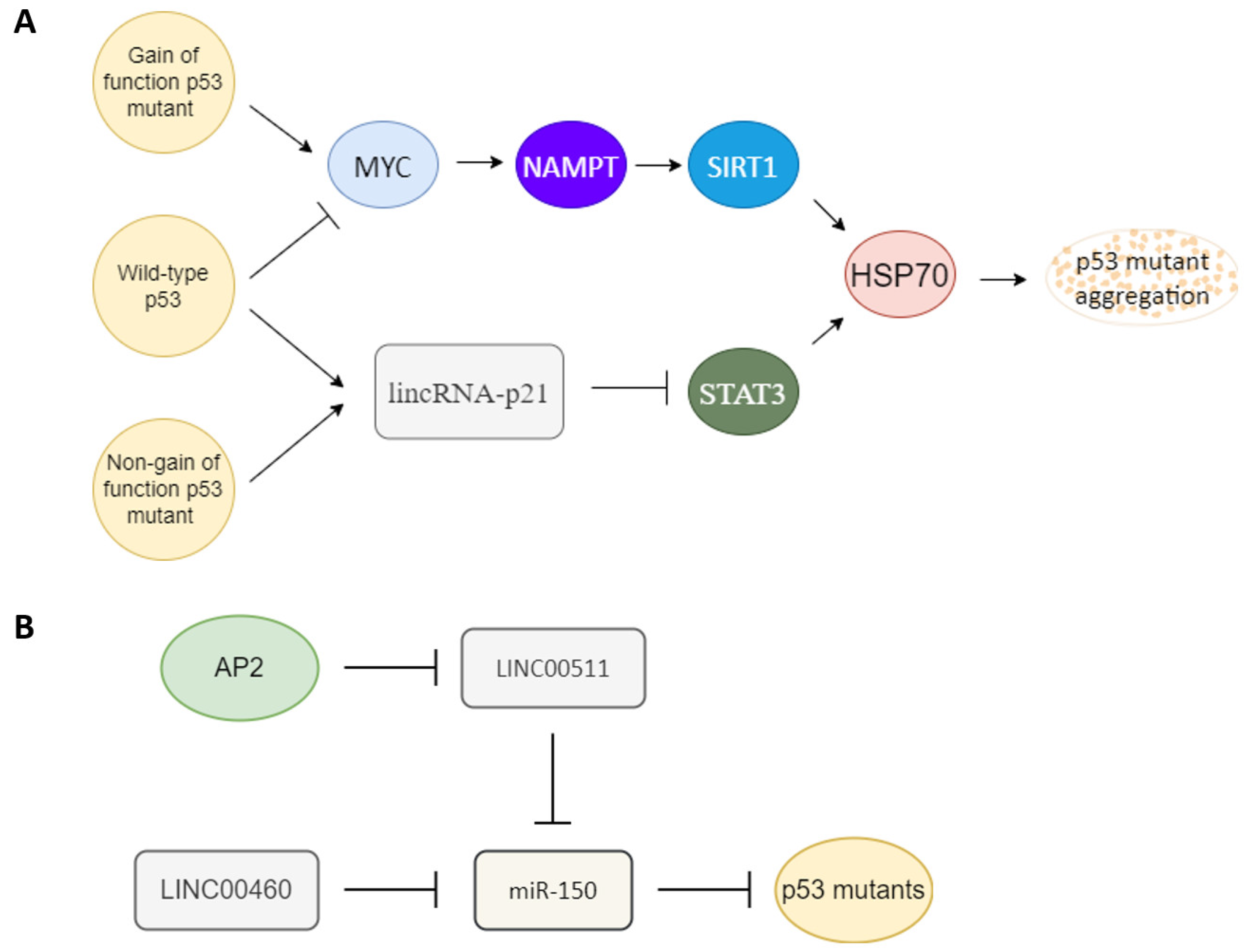
Figure 2. Key upstream molecules that influence p53 gain of function. (A) MYC and lincRNA-p21 are likely the key switch molecules that determine the gain-of-function or non-gain-of-function p53 signal to regulate the HSP70 expression to influence p53 mutant aggregation. Gain-of-function p53 mutant may induce the MYC-NAMPT- SIRT1-HSP70 axis to induce p53 auto-aggregation, and non-gain of function p53 mutants still can maintain non-aggregated p53 through the lincRNA-p21-STAT3-HSP70 axis. Wild-type p53 likely keeps its non-aggregated status through repression of MYC and upregulation of lincRNA-p21. (B) Oncogenic-type long-noncoding RNA LINC00511 and LINC00460 can act as a sponge to repress miR-150 to promote mutant p53 accumulation. A tumor-suppressor type transcription factor, AP2, can repress LINC00511 expression to decrease the amount of mutant p53.
MircoRNA and long-noncoding RNA are also key regulators of mutant p53. miR-150 can repress p53 [72][73][74]. An oncogenic type long-noncoding RNA LINC00460 can act as a sponge to repress miR-150 targeting to enhance mutant p53 expression [75]. Another tumor-suppressor type transcription factor, AP2, frequently interacts synergistically with p53 to activate downstream genes such as p21, CD82, and NEU4 [76][77][78][79]. AP2 can repress another oncogenic-type long-noncoding RNA, LINC00511 expression [80]. LINC00511 also can act as a sponge to repress miR-150 targeting [81]. So AP2 may also repress mutant p53 expression through the LINC00511-miR-150 axis (Figure 2B). AP2 has also been reported to decrease the amount of p53 [82].
References
- Scoumanne, A.; Harms, K.L.; Chen, X. Structural basis for gene activation by p53 family members. Cancer Biol. Ther. 2005, 4, 1178–1185.
- Harms, K.L.; Chen, X. The functional domains in p53 family proteins exhibit both common and distinct properties. Cell Death Differ. 2006, 13, 890–897.
- Zawacka-Pankau, J.E. The Role of p53 Family in Cancer. Cancers 2022, 14, 823.
- Levrero, M.; De Laurenzi, V.; Costanzo, A.; Gong, J.; Wang, J.Y.; Melino, G. The p53/p63/p73 family of transcription factors: Overlapping and distinct functions. J. Cell Sci. 2000, 113 Pt 10, 1661–1670.
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373.
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53.
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008.
- Hagiwara, K.; McMenamin, M.G.; Miura, K.; Harris, C.C. Mutational analysis of the p63/p73L/p51/p40/CUSP/KET gene in human cancer cell lines using intronic primers. Cancer Res. 1999, 59, 4165–4169.
- Schwartz, D.I.; Lindor, N.M.; Walsh-Vockley, C.; Roche, P.C.; Mai, M.; Smith, D.I.; Liu, W.; Couch, F.J. p73 mutations are not detected in sporadic and hereditary breast cancer. Breast Cancer Res. Treat. 1999, 58, 25–29.
- Han, S.; Semba, S.; Abe, T.; Makino, N.; Furukawa, T.; Fukushige, S.; Takahashi, H.; Sakurada, A.; Sato, M.; Shiiba, K.; et al. Infrequent somatic mutations of the p73 gene in various human cancers. Eur. J. Surg. Oncol. 1999, 25, 194–198.
- Soussi, T. The history of p53. A perfect example of the drawbacks of scientific paradigms. EMBO Rep. 2010, 11, 822–826.
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956.
- Cai, B.H.; Chao, C.F.; Huang, H.C.; Lee, H.Y.; Kannagi, R.; Chen, J.Y. Roles of p53 Family Structure and Function in Non-Canonical Response Element Binding and Activation. Int. J. Mol. Sci. 2019, 20, 3681.
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221.
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708.
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947.
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160.
- Malkin, D. Li-fraumeni syndrome. Genes Cancer 2011, 2, 475–484.
- Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Gripp, K.W.; Mirzaa, G.M.; Amemiya, A. GeneReviews; University of Washington: Seattle, WA, USA, 1993.
- Hisada, M.; Garber, J.E.; Fung, C.Y.; Fraumeni, J.F.; Li, F.P. Multiple primary cancers in families with Li-Fraumeni syndrome. J. Natl. Cancer Inst. 1998, 90, 606–611.
- Lepre, M.G.; Omar, S.I.; Grasso, G.; Morbiducci, U.; Deriu, M.A.; Tuszynski, J.A. Insights into the Effect of the G245S Single Point Mutation on the Structure of p53 and the Binding of the Protein to DNA. Molecules 2017, 22, 1358.
- Meneghetti, B.V.; Wilson, R.; Dias, C.K.; Cadore, N.A.; Klamt, F.; Zaha, A.; Ferreira, H.B.; Monteiro, K.M. p53 mutants G245S and R337H associated with the Li-Fraumeni syndrome regulate distinct metabolic pathways. Biochimie 2022, 198, 141–154.
- Ribeiro, R.C.; Sandrini, F.; Figueiredo, B.; Zambetti, G.P.; Michalkiewicz, E.; Lafferty, A.R.; DeLacerda, L.; Rabin, M.; Cadwell, C.; Sampaio, G.; et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci. USA 2001, 98, 9330–9335.
- Achatz, M.I.; Olivier, M.; Le Calvez, F.; Martel-Planche, G.; Lopes, A.; Rossi, B.M.; Ashton-Prolla, P.; Giugliani, R.; Palmero, E.I.; Vargas, F.R.; et al. The TP53 mutation, R337H, is associated with Li-Fraumeni and Li-Fraumeni-like syndromes in Brazilian families. Cancer Lett. 2007, 245, 96–102.
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968.
- Hug, N.; Longman, D.; Cáceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495.
- Floquet, C.; Deforges, J.; Rousset, J.P.; Bidou, L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011, 39, 3350–3362.
- Zhang, M.; Heldin, A.; Palomar-Siles, M.; Öhlin, S.; Bykov, V.J.N.; Wiman, K.G. Synergistic Rescue of Nonsense Mutant Tumor Suppressor p53 by Combination Treatment with Aminoglycosides and Mdm2 Inhibitors. Front. Oncol. 2017, 7, 323.
- Trzaska, C.; Amand, S.; Bailly, C.; Leroy, C.; Marchand, V.; Duvernois-Berthet, E.; Saliou, J.M.; Benhabiles, H.; Werkmeister, E.; Chassat, T.; et al. 2,6-Diaminopurine as a highly potent corrector of UGA nonsense mutations. Nat. Commun. 2020, 11, 1509.
- Ferguson, M.W.; Gerak, C.A.N.; Chow, C.C.T.; Rastelli, E.J.; Elmore, K.E.; Stahl, F.; Hosseini-Farahabadi, S.; Baradaran-Heravi, A.; Coltart, D.M.; Roberge, M. The antimalarial drug mefloquine enhances TP53 premature termination codon readthrough by aminoglycoside G418. PLoS ONE 2019, 14, e0216423.
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y.; et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598.
- Kerem, E.; Hirawat, S.; Armoni, S.; Yaakov, Y.; Shoseyov, D.; Cohen, M.; Nissim-Rafinia, M.; Blau, H.; Rivlin, J.; Aviram, M.; et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: A prospective phase II trial. Lancet 2008, 372, 719–727.
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547.
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513.
- Baradaran-Heravi, A.; Balgi, A.D.; Hosseini-Farahabadi, S.; Choi, K.; Has, C.; Roberge, M. Effect of small molecule eRF3 degraders on premature termination codon readthrough. Nucleic Acids Res. 2021, 49, 3692–3708.
- Nakamura, Y.; Futamura, M.; Kamino, H.; Yoshida, K.; Arakawa, H. Identification of p53-46F as a super p53 with an enhanced ability to induce p53-dependent apoptosis. Cancer Sci. 2006, 97, 633–641.
- Emamzadah, S.; Tropia, L.; Vincenti, I.; Falquet, B.; Halazonetis, T.D. Reversal of the DNA-binding-induced loop L1 conformational switch in an engineered human p53 protein. J. Mol. Biol. 2014, 426, 936–944.
- Koga, H.; Deppert, W. Identification of genomic DNA sequences bound by mutant p53 protein (Gly245-->Ser) in vivo. Oncogene 2000, 19, 4178–4183.
- Nichols, N.M.; Matthews, K.S. Human p53 phosphorylation mimic, S392E, increases nonspecific DNA affinity and thermal stability. Biochemistry 2002, 41, 170–178.
- Blagosklonny, M.V. Loss of function and p53 protein stabilization. Oncogene 1997, 15, 1889–1893.
- Cho, Y.; Gorina, S.; Jeffrey, P.D.; Pavletich, N.P. Crystal structure of a p53 tumor suppressor-DNA complex: Understanding tumorigenic mutations. Science 1994, 265, 346–355.
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604.
- Davison, T.S.; Vagner, C.; Kaghad, M.; Ayed, A.; Caput, D.; Arrowsmith, C.H. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J. Biol. Chem. 1999, 274, 18709–18714.
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell Biol. 2001, 21, 1874–1887.
- Chan, W.M.; Siu, W.Y.; Lau, A.; Poon, R.Y. How many mutant p53 molecules are needed to inactivate a tetramer? Mol. Cell Biol. 2004, 24, 3536–3551.
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218.
- Donzelli, S.; Biagioni, F.; Fausti, F.; Strano, S.; Fontemaggi, G.; Blandino, G. Oncogenomic Approaches in Exploring Gain of Function of Mutant p53. Curr. Genom. 2008, 9, 200–207.
- Liebl, M.C.; Hofmann, T.G. The Role of p53 Signaling in Colorectal Cancer. Cancers 2021, 13, 2125.
- Liao, P.; Zeng, S.X.; Zhou, X.; Chen, T.; Zhou, F.; Cao, B.; Jung, J.H.; Del Sal, G.; Luo, S.; Lu, H. Mutant p53 Gains Its Function via c-Myc Activation upon CDK4 Phosphorylation at Serine 249 and Consequent PIN1 Binding. Mol. Cell 2017, 68, 1134–1146.e1136.
- Stindt, M.H.; Muller, P.A.; Ludwig, R.L.; Kehrloesser, S.; Dötsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015, 34, 4300–4310.
- Yang-Hartwich, Y.; Soteras, M.G.; Lin, Z.P.; Holmberg, J.; Sumi, N.; Craveiro, V.; Liang, M.; Romanoff, E.; Bingham, J.; Garofalo, F.; et al. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene 2015, 34, 3605–3616.
- Kim, S.; An, S.S. Role of p53 isoforms and aggregations in cancer. Medicine 2016, 95, e3993.
- De Smet, F.; Saiz Rubio, M.; Hompes, D.; Naus, E.; De Baets, G.; Langenberg, T.; Hipp, M.S.; Houben, B.; Claes, F.; Charbonneau, S.; et al. Nuclear inclusion bodies of mutant and wild-type p53 in cancer: A hallmark of p53 inactivation and proteostasis remodelling by p53 aggregation. J. Pathol. 2017, 242, 24–38.
- Kanapathipillai, M. Treating p53 Mutant Aggregation-Associated Cancer. Cancers 2018, 10, 154.
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295.
- de Oliveira, G.A.P.; Petronilho, E.C.; Pedrote, M.M.; Marques, M.A.; Vieira, T.C.R.G.; Cino, E.A.; Silva, J.L. The Status of p53 Oligomeric and Aggregation States in Cancer. Biomolecules 2020, 10, 548.
- Pedrote, M.M.; Motta, M.F.; Ferretti, G.D.S.; Norberto, D.R.; Spohr, T.C.L.S.; Lima, F.R.S.; Gratton, E.; Silva, J.L.; de Oliveira, G.A.P. Oncogenic Gain of Function in Glioblastoma Is Linked to Mutant p53 Amyloid Oligomers. iScience 2020, 23, 100820.
- Boysen, M.; Kityk, R.; Mayer, M.P. Hsp70- and Hsp90-Mediated Regulation of the Conformation of p53 DNA Binding Domain and p53 Cancer Variants. Mol. Cell 2019, 74, 831–843.e4.
- Wu, H.; Dyson, H.J. Aggregation of zinc-free p53 is inhibited by Hsp90 but not other chaperones. Protein Sci. 2019, 28, 2020–2023.
- Westerheide, S.D.; Anckar, J.; Stevens, S.M.; Sistonen, L.; Morimoto, R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 2009, 323, 1063–1066.
- Zhang, T.; Berrocal, J.G.; Frizzell, K.M.; Gamble, M.J.; DuMond, M.E.; Krishnakumar, R.; Yang, T.; Sauve, A.A.; Kraus, W.L. Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J. Biol. Chem. 2009, 284, 20408–20417.
- Ganci, F.; Pulito, C.; Valsoni, S.; Sacconi, A.; Turco, C.; Vahabi, M.; Manciocco, V.; Mazza, E.M.C.; Meens, J.; Karamboulas, C.; et al. PI3K Inhibitors Curtail MYC-Dependent Mutant p53 Gain-of-Function in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2956–2971.
- Menssen, A.; Hydbring, P.; Kapelle, K.; Vervoorts, J.; Diebold, J.; Lüscher, B.; Larsson, L.G.; Hermeking, H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc. Natl. Acad. Sci. USA 2012, 109, E187–E196.
- Hermeking, H.; Lengauer, C.; Polyak, K.; He, T.C.; Zhang, L.; Thiagalingam, S.; Kinzler, K.W.; Vogelstein, B. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol. Cell 1997, 1, 3–11.
- Cai, B.H.; Chen, J.Y.; Lu, M.H.; Chang, L.T.; Lin, H.C.; Chang, Y.M.; Chao, C.F. Functional four-base A/T gap core sequence CATTAG of P53 response elements specifically bound tetrameric P53 differently than two-base A/T gap core sequence CATG bound both dimeric and tetrameric P53. Nucleic Acids Res. 2009, 37, 1984–1990.
- Phan, L.; Chou, P.C.; Velazquez-Torres, G.; Samudio, I.; Parreno, K.; Huang, Y.; Tseng, C.; Vu, T.; Gully, C.; Su, C.H.; et al. The cell cycle regulator 14-3-3σ opposes and reverses cancer metabolic reprogramming. Nat. Commun. 2015, 6, 7530.
- Petr, M.; Helma, R.; Polášková, A.; Krejčí, A.; Dvořáková, Z.; Kejnovská, I.; Navrátilová, L.; Adámik, M.; Vorlíčková, M.; Brázdová, M. Wild-type p53 binds to MYC promoter G-quadruplex. Biosci. Rep. 2016, 36, e00397.
- He, Y.H.; Yeh, M.H.; Chen, H.F.; Wang, T.S.; Wong, R.H.; Wei, Y.L.; Huynh, T.K.; Hu, D.W.; Cheng, F.J.; Chen, J.Y.; et al. ERα determines the chemo-resistant function of mutant p53 involving the switch between lincRNA-p21 and DDB2 expressions. Mol. Ther. Nucleic Acids 2021, 25, 536–553.
- Jin, S.; Yang, X.; Li, J.; Yang, W.; Ma, H.; Zhang, Z. p53-targeted lincRNA-p21 acts as a tumor suppressor by inhibiting JAK2/STAT3 signaling pathways in head and neck squamous cell carcinoma. Mol. Cancer 2019, 18, 38.
- Madamanchi, N.R.; Li, S.; Patterson, C.; Runge, M.S. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arter. Thromb. Vasc. Biol. 2001, 21, 321–326.
- Jego, G.; Hermetet, F.; Girodon, F.; Garrido, C. Chaperoning STAT3/5 by Heat Shock Proteins: Interest of Their Targeting in Cancer Therapy. Cancers 2019, 12, 21.
- Zhang, N.; Wei, X.; Xu, L. miR-150 promotes the proliferation of lung cancer cells by targeting P53. FEBS Lett. 2013, 587, 2346–2351.
- Wang, D.T.; Ma, Z.L.; Li, Y.L.; Wang, Y.Q.; Zhao, B.T.; Wei, J.L.; Qi, X.; Zhao, X.T.; Jin, Y.X. miR-150, p53 protein and relevant miRNAs consist of a regulatory network in NSCLC tumorigenesis. Oncol. Rep. 2013, 30, 492–498.
- Liu, F.; Di Wang, X. miR-150-5p represses TP53 tumor suppressor gene to promote proliferation of colon adenocarcinoma. Sci. Rep. 2019, 9, 6740.
- Meng, X.; Sun, W.; Yu, J.; Zhou, Y.; Gu, Y.; Han, J.; Zhou, L.; Jiang, X.; Wang, C. LINC00460-miR-149-5p/miR-150-5p-Mutant p53 Feedback Loop Promotes Oxaliplatin Resistance in Colorectal Cancer. Mol. Ther. Nucleic Acids 2020, 22, 1004–1015.
- McPherson, L.A.; Loktev, A.V.; Weigel, R.J. Tumor suppressor activity of AP2alpha mediated through a direct interaction with p53. J. Biol. Chem. 2002, 277, 45028–45033.
- Marreiros, A.; Dudgeon, K.; Dao, V.; Grimm, M.O.; Czolij, R.; Crossley, M.; Jackson, P. KAI1 promoter activity is dependent on p53, junB and AP2: Evidence for a possible mechanism underlying loss of KAI1 expression in cancer cells. Oncogene 2005, 24, 637–649.
- Tsai, Y.C.; Weissman, A.M. Dissecting the diverse functions of the metastasis suppressor CD82/KAI1. FEBS Lett. 2011, 585, 3166–3173.
- Cai, B.H.; Wu, P.H.; Chou, C.K.; Huang, H.C.; Chao, C.C.; Chung, H.Y.; Lee, H.Y.; Chen, J.Y.; Kannagi, R. Synergistic activation of the NEU4 promoter by p73 and AP2 in colon cancer cells. Sci. Rep. 2019, 9, 950.
- Quan, X.; Zhao, M.; Yang, X.; Zhu, Y.; Tian, X. AP2γ mediated downregulation of lncRNA LINC00511 as a ceRNA suppresses trophoblast invasion by regulating miR-29b-3p/Cyr61 axis. Biomed. Pharmacother. 2019, 120, 109269.
- Shi, G.; Cheng, Y.; Zhang, Y.; Guo, R.; Li, S.; Hong, X. Long non-coding RNA LINC00511/miR-150/MMP13 axis promotes breast cancer proliferation, migration and invasion. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165957.
- Stabach, P.R.; Thiyagarajan, M.M.; Woodfield, G.W.; Weigel, R.J. AP2alpha alters the transcriptional activity and stability of p53. Oncogene 2006, 25, 2148–2159.
More
Information
Subjects:
Medicine, Research & Experimental
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
3.8K
Revisions:
2 times
(View History)
Update Date:
08 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No