Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Bi-He Cai and Version 2 by Peter Tang.
The members of the p53 family comprise p53, p63, and p73, and full-length isoforms of the p53 family have a tumor suppressor function. However, p53, but not p63 or p73, has a high mutation rate in cancers causing it to lose its tumor suppressor function.
- p53
- mutation
- gain of function
- anti-cancer drugs
1. Introduction to the p53 Family
The p53 family has three members, p53, p63, and p73 [1][2][3][1,2,3]. TA (transactivation) isoforms of p53 family members are tumor suppressor genes [4][5][4,5]. p53 has a high frequency of mutation in cancers causing loss of its tumor suppression function [6][7][6,7]; however, p63 and p73 are rarely mutated in cancers [8][9][10][8,9,10].
1.1. p53
p53 was the second tumor suppressor gene identified, although p53 was actually discovered in 1979, before the first tumor suppressor gene Rb, which was cloned in 1986 [11][13]. p53 has a function in apoptosis, cell cycle arrest, autophagy, metabolism, DNA repair, translational control, and feedback mechanisms [12][13][14,15]. p53 knockout mice are prone to the spontaneous variety of tumors by 6 months of age (~age 34 in humans) [14][16]. p53 has an average ~50% mutation rate in cancers [6][7][15][6,7,17]. According to the Catalogue of Somatic Mutations in Cancer (COSMIC) database [16][18], there are 177,561 unique clinical samples with 50,215 unique samples having p53 mutations (Figure 1A). The top p53 mutation type is missense mutation accounting for 62.74% of mutations, and the second most prevalent mutation of p53 is a nonsense mutation which accounts for 10.72% of mutations. Eight missense mutations R175H, G245S, R248Q, R248W, R249S, R273H, R273S, and R282W called hotspot mutations account for ~28% of all p53 mutations identified in cancers [17][19]. p53 germline mutation can cause Li-Fraumeni syndrome (LFS) which is a hereditary syndrome with a relatively early age of cancer diagnosis usually before the age of 36 [18][20]. This syndrome is characterized by the early onset of various types of cancer such as soft-tissue, breast, brain, leukemia, lymphoma, gastrointestinal, head and neck, kidney, larynx, lung, skin, ovary, pancreas, prostate, testis, thyroid, and adrenocortical cancers [19][20][21,22]. Some congenital p53 mutations of the LFS are similar to those acquired by p53 hotspot mutations in cancers such as G245S [21][22][23,24], but some congenital p53 mutations only appear in LFS as germ-line specific mutations such as R337H [23][24][25,26].
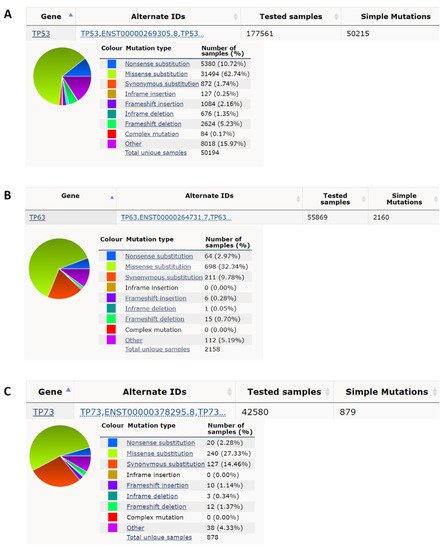
Figure 1. p53 has a high nonsense and missense mutation rate compared to p63 and p73 in cancer. (A) p53 has, respectively, a 10.72% and 62.74% nonsense mutation rate and missense mutation rate in all cancer mutation samples from the COSMIC database (https://cancer.sanger.ac.uk/cosmic; accessed date (29 April 2022) [16][18]. (B) p63 has, respectively, 2.97% and 32.34% nonsense mutation rate and missense mutation rate in all cancer mutation samples. (C) p73 has, respectively, 2.28% and 27.33%, nonsense mutation rate and missense mutation rate in all cancer mutation samples.
2. Types of p53 Mutations in Cancers
2.1. Nonsense Mutation
p53 nonsense mutations comprise ~10% of all p53 mutants (Figure 1A); the actual p53 nonsense mutation rate is higher than the average ~5% [25][38]. There are three pre-stop DNA codons, TAA, TAG, and TGA. Nonsense mutation leads to the generation of premature termination codons (PTC), which leads to nonsense-mediated mRNA decay (NMD), resulting in the inability to express full-length proteins and extremely low expression levels of truncated proteins [26][39]. Two mechanisms are known to regenerate full-length proteins, one is to inhibit NMD, and the other is for PTC readthrough. It is known that aminoglycoside drugs, such as G418 and gentamicin, can inhibit NMD and promote p53 PTC readthrough [27][28][40,41]. But these drugs are highly toxic and cannot be used in clinical practice. 2,6-Diaminopurine (DAP), can inhibit the activity of putative ribosomal RNA methyltransferase 1 (FTSJ1) to increase the capacity of tRNATrp to recognize the UGA stop codon to promote p53 PTC readthrough, but this drug does not have the ability to inhibit NMD [29][42]. Furthermore, DAP is only effective for nonsense mutations of TGA but not TAA or TAG [29][42], and this greatly reduces the available targets. In addition, some phthalimide derivatives and antimalarial drug quinines can promote the p53 PTC read-through ability of G418 to increase the proportion of full-length p53 and to reduce the expression of the truncated protein, but these drugs alone have no effect on PTC read-through [30][31][43,44]. Non-aminoglycoside drugs, such as Ataluren (PTC124), also increase the read-through ability of PTC without the ability to inhibit NMD, and PTC124 has been used in clinical phase II or III trials to treat genetic diseases with specific nonsense mutations [32][33][45,46]. PTC124 can also promote p53 PTC readthrough [34][47]. A recent study has shown that CC-885 and CC-90009 can inhibit NMD; of note, the effective concentration of CC-885 for treatment of p53 nonsense mutation with TAA is only one-tenth of that of CC-90009 [35][48].
2.2. Loss-of-Function Mutants
p53 missense mutations contain both loss of function and gain of function. p53 mutation is mainly located at the N-terminal transactivation domain or middle DNA binding domain [2]. In addition, several point mutants still have normal DNA binding function [36][37][38][39][49,50,51,52]; most p53 mutations within the N-terminal transactivation domain or the DNA binding domain lose their transactivation function or DNA binding function causing loss of their tumor suppressor functions such as cell cycle checkpoint controls and apoptosis [40][41][42][53,54,55]. These p53 mutations can associate with p63 and p73, whereas wild-type p53 cannot [43][44][56,57]. Therefore, loss-of-function p53 mutants act in the same way as the ∆N isofroms of the p53 family having a dominant-negative effect to repress the functions of normal TA isoforms of p53 family members [44][45][57,58].
2.3. Gain-of-Function Mutations
Some p53 mutants can obtain some oncogenic functions such as cell migration, invasion, and metastasis to enhance tumorigenesis [46][59], and these p53 mutants are called gain-of-function mutants. The acquisition of p53 gain-of-function is via three mechanisms [47][48][60,61]. First, mutant p53 can directly bind to the novel binding site with a p53 non-canonical sequence to activate several oncogenic genes [38][51]. Second, mutant p53 can act as a co-activator to bind to other transcription factors to activate some oncogenic genes [49][62]. Third, mutant p53 can bind to other tumor suppressive-type transcription factors to cause loss of transcription ability [50][63]. Some p53 mutants can become aggregated in several types of cancer, such as breast, lung ovary, colorectal, and head and neck cancers [51][52][53][54][64,65,66,67]. It is known that these aggregations of mutant p53 can sequester other tumor suppressor genes as a third mechanism to cause p53 gain of function [55][56][57][68,69,70].
3. Factors Influencing p53 Mutant Gain of Function
Several factors have been reported to influence p53 mutant gain of function (Figure 2A). The heat shock protein 70 (HSP70) has been reported to enhance mutant p53 aggregation [58][71], and heat shock protein 90 (HSP90) can repress mutant p53 aggregation [58][59][71,72]. SIRT1 is an NAD+ dependent histone deacetylase that has been reported to deacetylate HSF1 to enhance HSF1 transcriptional activity to increase HSP70 expression [60][73]. NAMPT can enhance SIRT1 activity by increasing the amount of NAD+ [61][74]. p53 gain-of-function mutant can induce MYC [49][62][62,75], and MYC can enhance NAPMT [63][76]. Wild-type p53 can induce 14-3-3σ expression [64][65][77,78], and 14-3-3σ can promote MYC poly-ubiquitination and degradation [66][79]. Another article also reported that wild-type p53 can bind to G-quadruplexes on MYC promoter to repress MYC expression [67][80]. Both wild-type and non-gain-of-function p53 mutants can activate lincRNA-p21 through binding to G-quadruplexes on lincRNA-p21 promoter [68][81], and lincRNA-p21 can repress STAT3 [69][82]. STAT3 can bind to the HSP70 promoter to active HSP70 expression [70][71][83,84]. The relationships between factors that influence p53 mutant gain of function are summarized in Figure 2A.
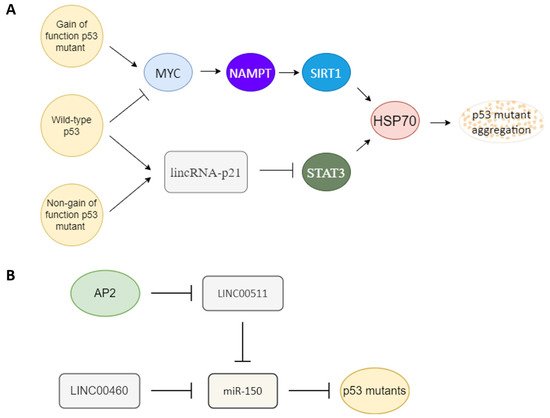
Figure 2. Key upstream molecules that influence p53 gain of function. (A) MYC and lincRNA-p21 are likely the key switch molecules that determine the gain-of-function or non-gain-of-function p53 signal to regulate the HSP70 expression to influence p53 mutant aggregation. Gain-of-function p53 mutant may induce the MYC-NAMPT- SIRT1-HSP70 axis to induce p53 auto-aggregation, and non-gain of function p53 mutants still can maintain non-aggregated p53 through the lincRNA-p21-STAT3-HSP70 axis. Wild-type p53 likely keeps its non-aggregated status through repression of MYC and upregulation of lincRNA-p21. (B) Oncogenic-type long-noncoding RNA LINC00511 and LINC00460 can act as a sponge to repress miR-150 to promote mutant p53 accumulation. A tumor-suppressor type transcription factor, AP2, can repress LINC00511 expression to decrease the amount of mutant p53.
MircoRNA and long-noncoding RNA are also key regulators of mutant p53. miR-150 can repress p53 [72][73][74][85,86,87]. An oncogenic type long-noncoding RNA LINC00460 can act as a sponge to repress miR-150 targeting to enhance mutant p53 expression [75][88]. Another tumor-suppressor type transcription factor, AP2, frequently interacts synergistically with p53 to activate downstream genes such as p21, CD82, and NEU4 [76][77][78][79][89,90,91,92]. AP2 can repress another oncogenic-type long-noncoding RNA, LINC00511 expression [80][93]. LINC00511 also can act as a sponge to repress miR-150 targeting [81][94]. So AP2 may also repress mutant p53 expression through the LINC00511-miR-150 axis (Figure 2B). AP2 has also been reported to decrease the amount of p53 [82][95].