 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Annamaria Aprile | -- | 3293 | 2022-07-06 20:03:45 | | | |
| 2 | Jessie Wu | + 4 word(s) | 3297 | 2022-07-07 05:26:38 | | |
Video Upload Options
Hemoglobinopathies are inherited disorders affecting hemoglobin (Hb) production, estimated to be the most common monogenic diseases worldwide. In the last decade, research on pathophysiology and therapeutic solutions for β-thalassemia (BThal) and sickle cell disease (SCD) has been mostly focused on the primary erythroid defect, thus neglecting the study of hematopoietic stem cells (HSCs) and bone marrow (BM) microenvironment. The quality and engraftment of HSCs depend on the BM microenvironment, influencing the outcome of HSC transplantation (HSCT) both in allogeneic and in autologous gene therapy settings. In BThal and SCD, the consequences of severe anemia alter erythropoiesis and cause chronic stress in different organs, including the BM.
1. Introduction
2. Hematopoietic Stem Cells
3. The Stromal Niche
4. Hematopoietic and Soluble Niche Factors
5. The Role of IO in the β-Thalassemia Bone Marrow Niche
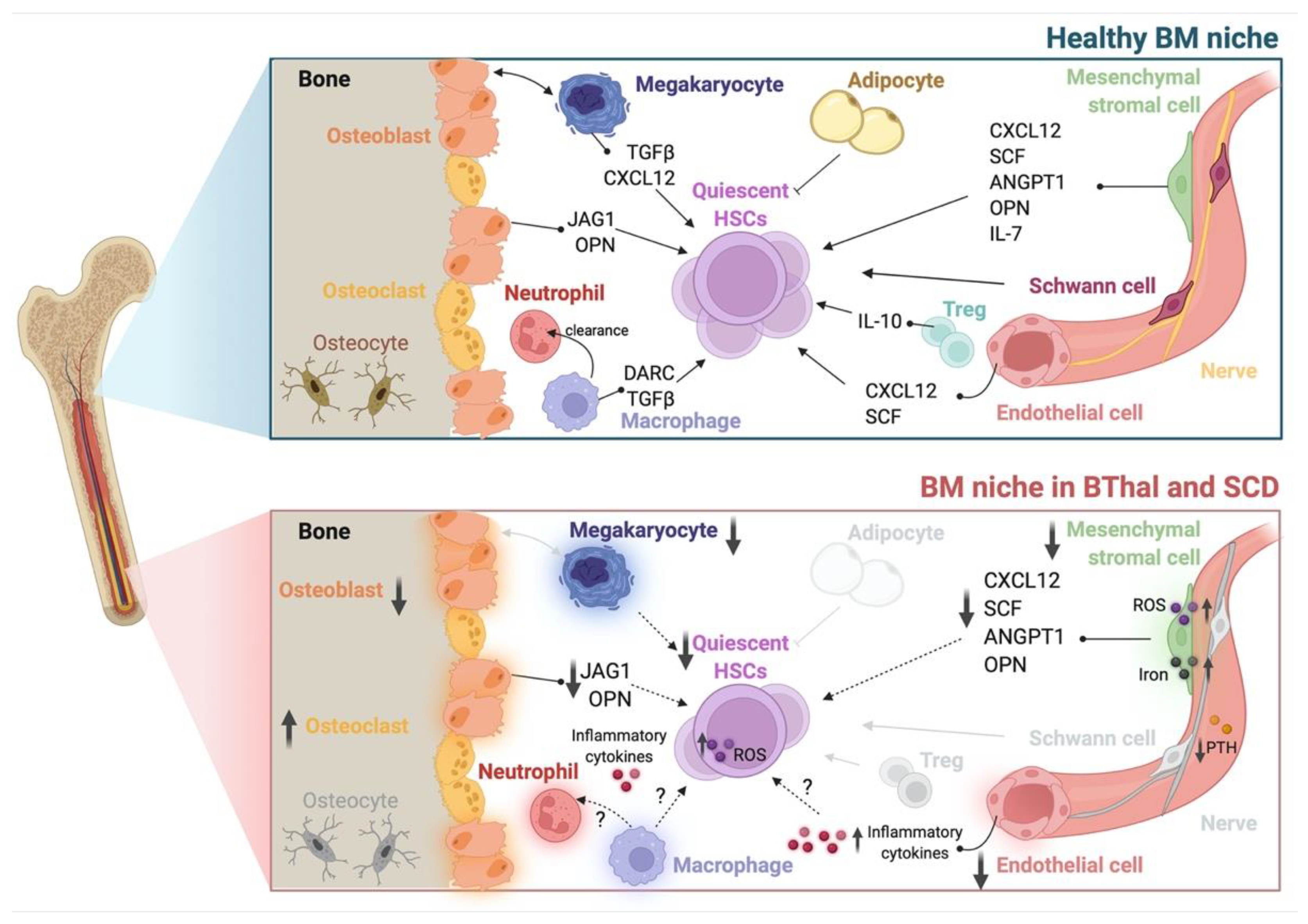
| Cell Population | Disease | Species | Alterations | References |
|---|---|---|---|---|
| HSC | BThal | mouse | ⇓ number ⇓ quiescence ⇓ stemness ⇓ reconstitution capacity ⇑ response to stress |
[2] |
| human | ⇓ frequency ⇓ quiescence ⇓ stemness ⇑ response to stress (HSPC) |
[2][9][10] | ||
| SCD | mouse | ⇓ frequency ⇑ ROS ⇑ DNA damage ⇓ quiescence (HSPC) ⇑ mobilization (HSPC) |
[3][11][12] | |
| human | ⇓ frequency ⇑ mobilization |
[9][13] | ||
| Osteolineage cell | BThal | mouse | ⇓ BMD ⇓ systemic PTH ⇓ OB activity ⇓ niche molecules ⇑ FGF23 |
[2][15][29] |
| human | ⇓ niche molecules | [2] | ||
| SCD | mouse | ⇓ bone microarchitecture | [24] | |
| ⇑ osteoclastogenesis ⇓ osteogenic factors |
[25] | |||
| MSC | BThal | mouse | ⇓ frequency ⇓ niche molecules |
[2] |
| human | ⇓ frequency ⇓ osteogenic and adipogenic potential ⇑ ROS ⇑ iron content ⇓ niche molecules ⇓ HSPC maintenance |
[18] | ||
| SCD | mouse | ⇓ frequency ⇑ ROS ⇓ osteogenic and adipogenic potential ⇓ niche molecules ⇓ HSC maintenance |
[3] | |
| human | ⇓ niche molecules | [27] | ||
| EC | SCD | mouse | altered BM vasculature ⇑ inflammatory cytokines |
[28] |
| MK | BThal | mouse | ⇓ systemic TPO ⇓ maturation ⇓ niche molecules |
[32] |
| Neutrophil | BThal | mouse | altered maturation | [38] |
References
- Cavazzana, M.; Ribeil, J.A.; Lagresle-Peyrou, C.; Andre-Schmutz, I. Gene Therapy with Hematopoietic Stem Cells: The Diseased Bone Marrow’s Point of View. Stem Cells Dev. 2017, 26, 71–76.
- Aprile, A.; Gulino, A.; Storto, M.; Villa, I.; Beretta, S.; Merelli, I.; Rubinacci, A.; Ponzoni, M.; Marktel, S.; Tripodo, C.; et al. Hematopoietic stem cell function in beta-thalassemia is impaired and is rescued by targeting the bone marrow niche. Blood 2020, 136, 610–622.
- Tang, A.; Strat, A.N.; Rahman, M.; Zhang, H.; Bao, W.; Liu, Y.; Shi, D.; An, X.; Manwani, D.; Shi, P.; et al. Murine bone marrow mesenchymal stromal cells have reduced hematopoietic maintenance ability in sickle cell disease. Blood 2021, 138, 2570–2582.
- McColl, B.; Vadolas, J. Animal models of beta-hemoglobinopathies: Utility and limitations. J. Blood Med. 2016, 7, 263–274.
- Yang, B.; Kirby, S.; Lewis, J.; Detloff, P.J.; Maeda, N.; Smithies, O. A mouse model for beta 0-thalassemia. Proc. Natl. Acad. Sci. USA 1995, 92, 11608–11612.
- Trudel, M.; Saadane, N.; Garel, M.C.; Bardakdjian-Michau, J.; Blouquit, Y.; Guerquin-Kern, J.L.; Rouyer-Fessard, P.; Vidaud, D.; Pachnis, A.; Romeo, P.H.; et al. Towards a transgenic mouse model of sickle cell disease: Hemoglobin SAD. EMBO J. 1991, 10, 3157–3165.
- Paszty, C.; Brion, C.M.; Manci, E.; Witkowska, H.E.; Stevens, M.E.; Mohandas, N.; Rubin, E.M. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science 1997, 278, 876–878.
- Ryan, T.M.; Ciavatta, D.J.; Townes, T.M. Knockout-transgenic mouse model of sickle cell disease. Science 1997, 278, 873–876.
- Hua, P.; Roy, N.; de la Fuente, J.; Wang, G.; Thongjuea, S.; Clark, K.; Roy, A.; Psaila, B.; Ashley, N.; Harrington, Y.; et al. Single-cell analysis of bone marrow-derived CD34+ cells from children with sickle cell disease and thalassemia. Blood 2019, 134, 2111–2115.
- Lidonnici, M.R.; Chianella, G.; Tiboni, F.; Barcella, M.; Merelli, I.; Scaramuzza, S.; Rossi, C.; Crippa, S.; Storto, M.; Bernardo, M.E.; et al. S269 TGF-beta signaling controls the lineage cell fate of hematopoietic stem cells towards erythroid branching in beta-thalassemia. EHA2021 Virtual Congress Abstract Book. HemaSphere 2021, 5, e566.
- Javazon, E.H.; Radhi, M.; Gangadharan, B.; Perry, J.; Archer, D.R. Hematopoietic stem cell function in a murine model of sickle cell disease. Anemia 2012, 2012, 387385.
- Blouin, M.J.; De Paepe, M.E.; Trudel, M. Altered hematopoiesis in murine sickle cell disease. Blood 1999, 94, 1451–1459.
- Tolu, S.S.; Wang, K.; Yan, Z.; Zhang, S.; Roberts, K.; Crouch, A.S.; Sebastian, G.; Chaitowitz, M.; Fornari, E.D.; Schwechter, E.M.; et al. Characterization of Hematopoiesis in Sickle Cell Disease by Prospective Isolation of Stem and Progenitor Cells. Cells 2020, 9, 2159.
- Rachmilewitz, E.A.; Giardina, P.J. How I treat thalassemia. Blood 2011, 118, 3479–3488.
- Vogiatzi, M.G.; Tsay, J.; Verdelis, K.; Rivella, S.; Grady, R.W.; Doty, S.; Giardina, P.J.; Boskey, A.L. Changes in bone microarchitecture and biomechanical properties in the th3 thalassemia mouse are associated with decreased bone turnover and occur during the period of bone accrual. Calcif. Tissue Int. 2010, 86, 484–494.
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841–846.
- Nilsson, S.K.; Johnston, H.M.; Whitty, G.A.; Williams, B.; Webb, R.J.; Denhardt, D.T.; Bertoncello, I.; Bendall, L.J.; Simmons, P.J.; Haylock, D.N. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood 2005, 106, 1232–1239.
- Crippa, S.; Rossella, V.; Aprile, A.; Silvestri, L.; Rivis, S.; Scaramuzza, S.; Pirroni, S.; Avanzini, M.A.; Basso-Ricci, L.; Hernandez, R.J.; et al. Bone marrow stromal cells from β-thalassemia patients have impaired hematopoietic supportive capacity. J. Clin. Investig. 2019, 129, 1566–1580.
- Almeida, A.; Roberts, I. Bone involvement in sickle cell disease. Br. J. Haematol. 2005, 129, 482–490.
- Baldwin, C.; Nolan, V.G.; Wyszynski, D.F.; Ma, Q.L.; Sebastiani, P.; Embury, S.H.; Bisbee, A.; Farrell, J.; Farrer, L.; Steinberg, M.H. Association of klotho, bone morphogenic protein 6, and annexin A2 polymorphisms with sickle cell osteonecrosis. Blood 2005, 106, 372–375.
- Seguin, C.; Kassis, J.; Busque, L.; Bestawros, A.; Theodoropoulos, J.; Alonso, M.L.; Harvey, E.J. Non-traumatic necrosis of bone (osteonecrosis) is associated with endothelial cell activation but not thrombophilia. Rheumatology 2008, 47, 1151–1155.
- Nouraie, M.; Cheng, K.; Niu, X.; Moore-King, E.; Fadojutimi-Akinsi, M.F.; Minniti, C.P.; Sable, C.; Rana, S.; Dham, N.; Campbell, A.; et al. Predictors of osteoclast activity in patients with sickle cell disease. Haematologica 2011, 96, 1092–1098.
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J. Exp. Med. 2000, 191, 275–286.
- Green, M.; Akinsami, I.; Lin, A.; Banton, S.; Ghosh, S.; Chen, B.; Platt, M.; Osunkwo, I.; Ofori-Acquah, S.; Guldberg, R.; et al. Microarchitectural and mechanical characterization of the sickle bone. J. Mech. Behav. Biomed. Mater. 2015, 48, 220–228.
- Dalle Carbonare, L.; Matte, A.; Valenti, M.T.; Siciliano, A.; Mori, A.; Schweiger, V.; Zampieri, G.; Perbellini, L.; De Franceschi, L. Hypoxia-reperfusion affects osteogenic lineage and promotes sickle cell bone disease. Blood 2015, 126, 2320–2328.
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589.
- Stenger, E.O.; Chinnadurai, R.; Yuan, S.; Garcia, M.; Arafat, D.; Gibson, G.; Krishnamurti, L.; Galipeau, J. Bone Marrow-Derived Mesenchymal Stromal Cells from Patients with Sickle Cell Disease Display Intact Functionality. Biol. Blood Marrow Transplant. 2017, 23, 736–745.
- Park, S.Y.; Matte, A.; Jung, Y.; Ryu, J.; Anand, W.B.; Han, E.Y.; Liu, M.; Carbone, C.; Melisi, D.; Nagasawa, T.; et al. Pathologic angiogenesis in the bone marrow of humanized sickle cell mice is reversed by blood transfusion. Blood 2020, 135, 2071–2084.
- Aprile, A.; Raggi, L.; Storto, M.; Villa, I.; Bolamperti, S.; Marktel, S.; Motta, I.; Cappellini, M.D.; Rubinacci, A.; Ferrari, G. Inhibition of Fibroblast Growth Factor-23 (FGF-23) Rescues Bone and Hematopoietic Stem Cell Niche Defects in Beta-Thalassemia, Uncovering the Missing Link Between Hematopoiesis and Bone. Blood 2021, 138 (Suppl. 1), 572.
- Edmonston, D.; Wolf, M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19.
- Clinkenbeard, E.L.; Hanudel, M.R.; Stayrook, K.R.; Appaiah, H.N.; Farrow, E.G.; Cass, T.A.; Summers, L.J.; Ip, C.S.; Hum, J.M.; Thomas, J.C.; et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica 2017, 102, e427–e430.
- Aprile, A.; Storto, M.; Malara, A.; Gulino, A.; Raggi, L.; Sighinolfi, S.; Beretta, S.; Merelli, I.; Marktel, S.; Ponzoni, M.; et al. S249 Reduced Levels of thrombopoietin Contribute to Impaired Hematopoietic stem Cell Function and Defective Megakaryopoiesis in Beta-Thalassemia. Available online: https://library.ehaweb.org/eha/2021/eha2021-virtual-congress/324657/annamaria.aprile.reduced.levels.of.thrombopoietin.contribute.to.impaired.html?f=menu%3D6%2Abrowseby%3D8%2Asortby%3D2%2Amedia%3D3%2Ace_id%3D2035%2Aot_id%3D25563 (accessed on 4 May 2022).
- Decker, M.; Leslie, J.; Liu, Q.; Ding, L. Hepatic thrombopoietin is required for bone marrow hematopoietic stem cell maintenance. Science 2018, 360, 106–110.
- Yoshihara, H.; Arai, F.; Hosokawa, K.; Hagiwara, T.; Takubo, K.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Matsuoka, S.; Miyamoto, K.; et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 2007, 1, 685–697.
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320.
- Nakamura-Ishizu, A.; Takubo, K.; Kobayashi, H.; Suzuki-Inoue, K.; Suda, T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J. Exp. Med. 2015, 212, 2133–2146.
- Seyfried, A.N.; Maloney, J.M.; MacNamara, K.C. Macrophages Orchestrate Hematopoietic Programs and Regulate HSC Function During Inflammatory Stress. Front. Immunol. 2020, 11, 1499.
- Siwaponanan, P.; Siegers, J.Y.; Ghazali, R.; Ng, T.; McColl, B.; Ng, G.Z.; Sutton, P.; Wang, N.; Ooi, I.; Thiengtavor, C.; et al. Reduced PU.1 expression underlies aberrant neutrophil maturation and function in beta-thalassemia mice and patients. Blood 2017, 129, 3087–3099.
- Casanova-Acebes, M.; Pitaval, C.; Weiss, L.A.; Nombela-Arrieta, C.; Chevre, R.; A-González, N.; Kunisaki, Y.; Zhang, D.; van Rooijen, N.; Silberstein, L.E.; et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 2013, 153, 1025–1035.
- Zhang, D.; Xu, C.; Manwani, D.; Frenette, P.S. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 2016, 127, 801–809.
- Canalli, A.A.; Franco-Penteado, C.F.; Saad, S.T.; Conran, N.; Costa, F.F. Increased adhesive properties of neutrophils in sickle cell disease may be reversed by pharmacological nitric oxide donation. Haematologica 2008, 93, 605–609.
- Lum, A.F.; Wun, T.; Staunton, D.; Simon, S.I. Inflammatory potential of neutrophils detected in sickle cell disease. Am. J. Hematol. 2004, 76, 126–133.
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827.
- Zhang, D.; Chen, G.; Manwani, D.; Mortha, A.; Xu, C.; Faith, J.J.; Burk, R.D.; Kunisaki, Y.; Jang, J.E.; Scheiermann, C.; et al. Neutrophil ageing is regulated by the microbiome. Nature 2015, 525, 528–532.
- Villagra, J.; Shiva, S.; Hunter, L.A.; Machado, R.F.; Gladwin, M.T.; Kato, G.J. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 2007, 110, 2166–2172.
- Belcher, J.D.; Marker, P.H.; Weber, J.P.; Hebbel, R.P.; Vercellotti, G.M. Activated monocytes in sickle cell disease: Potential role in the activation of vascular endothelium and vaso-occlusion. Blood 2000, 96, 2451–2459.
- Mitroulis, I.; Kalafati, L.; Bornhauser, M.; Hajishengallis, G.; Chavakis, T. Regulation of the Bone Marrow Niche by Inflammation. Front. Immunol 2020, 11, 1540.
- Essers, M.A.; Offner, S.; Blanco-Bose, W.E.; Waibler, Z.; Kalinke, U.; Duchosal, M.A.; Trumpp, A. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009, 458, 904–908.
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486.
- Luo, Y.; Shao, L.; Chang, J.; Feng, W.; Liu, Y.L.; Cottler-Fox, M.H.; Emanuel, P.D.; Hauer-Jensen, M.; Bernstein, I.D.; Liu, L.; et al. M1 and M2 macrophages differentially regulate hematopoietic stem cell self-renewal and ex vivo expansion. Blood Adv. 2018, 2, 859–870.
- Zhou, X.; Huang, L.; Wu, J.; Qu, Y.; Jiang, H.; Zhang, J.; Qiu, S.; Liao, C.; Xu, X.; Xia, J.; et al. Impaired bone marrow microenvironment and stem cells in transfusion-dependent beta-thalassemia. Biomed. Pharmacother. 2022, 146, 112548.

