1. Introduction
The hematopoietic stem cell (HSC) bone marrow niche is still poorly investigated in hemoglobinopathies. The status of HSCs and BM niche, indeed, should be considered in the context of allogeneic HSCT to ensure a sustained engraftment of donor cells and especially in recent autologous gene therapy settings, to develop optimized protocols for increasing the efficacy and safety of ex-vivo genetic manipulation [
44]. The reduced number of HSCs available for collection and the impaired engraftment potential of HSCs in BThal and SCD mouse models [
114,
115] highlighted the need for a better characterization of the primitive HSC compartment and BM niche features to develop targeting strategies for improving the outcome of allogeneic and autologous HSCT.
To study HSC niche in BThal and SCD, mouse strains recapitulating the main features of the human diseases were exploited [
116]. An investigation of BThal HSCs and BM niche was performed in the Hbb
th3/+ (th3) murine model. th3 mice lack both the adult β
maj- and β
min-globin genes [
117]. Although homozygous knockout mice are not viable, heterozygotes survive with features of severe BThal intermedia, including reduced RBC and Hb concentrations, microcytosis, reduced hematocrit and elevated reticulocytes, IE, splenomegaly, bone malformation, and IO in multiple tissues. The studied mouse models for SCD include the transgenic SAD, Berkley, and Townes strains [
116]. They were generated by co-expression of the human α2-globin gene and a modified βS-globin gene, both linked to the β-globin locus regulatory region. The SAD mouse incorporates the βS variant with additional mutations known to enhance the severity of the sickle phenotype into a BThal background [
118], whereas the Berkley and Townes transgenic strains carry the genes for human α- and βS-globin on a genetic background deficient for the murine β-globin genes and incorporate a YAC containing one or both γ-globin gene sequences to avert the gestational lethality [
119,
120]. These models reproduce in vivo sickling of RBCs, hypoxia, and severe anemia with reduced hematocrit and increased reticulocytes, systemic microvascular occlusions, hemolytic and renal complications, splenomegaly, and IE of human SCD. Patient-derived samples were studied for the immunophenotypic characterization of HSCs and BM microenvironment to validate findings obtained in mice and to model in vitro the discovered alterations.
2. Hematopoietic Stem Cells
BThal. A recent study by Aprile et al. has provided the first demonstration of impaired HSC function caused by an altered BM niche in BThal [
114]. The authors demonstrated that th3 mice have a decreased number of HSCs, as compared to wild-type (wt) controls. Cell cycle analysis revealed a loss of quiescence with a lower frequency of HSCs in the G0 phase and an increased cycling rate with a higher fraction of cells accumulated in the S phase. These data were corroborated by RNA-seq experiments performed on sorted th3 HSCs, revealing a positive enrichment of cell-cycle-associated categories, a upregulation of genes involved in DNA damage, cellular responses to stress, and a downregulation of stemness genes (including
Cdkn1c,
Runx1l1,
Fgd5, and
Hes1), thus highlighting the increased replication stress and impaired self-renewal ability of HSCs in BThal. To evaluate the functional activity of BThal HSCs, they performed long-term in vivo transplant experiments and they observed a competitive disadvantage of th3 HSCs compared to wt ones when transplanted into th3 mice. Notably, transplantation into wt recipients rescued the long-term repopulating capacity of th3 HSCs, suggesting that the wt BM microenvironment had a corrective role in restoring HSC functions. Secondary transplants into wt animals showed that th3 HSCs recovered their reconstitution capacity with complete normalization of the quiescent state. On the contrary, th3 HSCs in BThal recipients underwent exhaustion over time. These results suggested that impaired HSC self-renewal and quiescence in BThal are not intrinsic defects, but their behavior is affected by prolonged residence in an altered BM microenvironment, which is progressively worsened by the disease.
In line with these findings, patients affected by TDT showed reduced quiescence of CD34
+CD38
- primitive HSPCs [
114]. Moreover, the gene expression profile of patients’ CD34
+ cells revealed an upregulation of genes associated with stress stimuli and DNA damage, thus indicating an impairment of HSPCs also in the human disease. Consistently, Hua and colleagues published a reduced frequency of Lin
− CD10
− CD34
+ CD38
-\lowCD45RA
− CD90
+ HSCs in the CD34
+ cell compartment of BThal pediatric patients [
121]. Moreover, and most importantly, RNA-seq analysis of BThal patients’ HSCs showed increased proliferation and reduced stemness [
122].
Overall, this evidence highlighted the importance of the BM microenvironment in preserving HSC fitness in the BThal context.
SCD. Studies in the murine models of SCD revealed defects in SCD hematopoiesis and HSCs. In SCD, the BM environment is highly enriched for ROS, mainly generated by SS-RBCs and the activated endothelium. By examining the effects of oxidative stress on SCD HSCs, Javazon et al. showed that SCD BM has a decreased colony forming unit potential and a reduced number of Lineage
- Sca1
+ cKit
+ (LSK) HSPCs [
123]. Cell cycle analyses revealed that fewer LSK cells were in the G0 phase, and a significant increase in lipid peroxidation and ROS in SCD LSKs was detected. HSPCs from SCD mice showed an impaired engraftment potential, which is partially restored by n-acetyl cysteine (NAC) antioxidant treatment of LSK cells before transplantation, thus suggesting that an altered redox environment in SCD affects HSC function.
The results of reduced clonogenic potential of SCD HSPCs are paralleled by the increased mobilization of multipotent cells in both mice and humans affected by SCD. A hematopoietic compensatory mechanism was described in SCD, consisting in the mobilization of progenitor cells from the BM to the peripheral blood and their subsequent uptake into the splenic extramedullary hematopoietic site in response to the erythropoietic stress [
124]. The spleen of SCD mice indeed contains significantly increased numbers of cycling erythroid colony-forming cells, indicating the strong proliferative pressure on the erythroid lineage.
Alterations in SCD HSCs were reported by Tang et al., who showed a decrease in HSC frequency, increased DNA damage, and an accumulation of ROS in HSCs from SCD mice, associated with the reduced hematopoietic supportive ability of MSCs [
115].
Recently, Hua et al. showed a lower proportion of Lin
− CD10
− CD34
+ CD38
-\lowCD45RA
- CD90
+ HSCs in the CD34
+ cell compartment of SCD pediatric patients, along with an increased frequency of CD34
+ CD10
+ lymphoid progenitor cells [
121], suggesting hematopoietic defects also in the human disease. Moreover, the characterization of circulating hematopoietic populations from adult and pediatric SCD patients confirmed an increase in CD34
bright HSPCs and the mobilization of primitive HSCs in the peripheral blood as compared to healthy controls [
125].
3. The Stromal Niche
BThal. To explain the defects in the transplantation outcome of th3 HSCs into a BThal BM niche, Aprile et al. focused their attention on the interactions between HSCs and stromal cells, such as osteolineage cells and MSCs in the BM of BThal mice [
114]. Bone disease is a common and severe complication of BThal, resulting from hormonal deficiency, BM expansion, and iron toxicity. Consistent with the common finding of osteoporosis and hypoparathyroidism in BThal patients [
27], data in th3 mice confirmed a reduced BMD [
126] caused by decreased OB activity and low levels of circulating PTH [
114]. Moreover, the authors showed reduced levels of key niche molecules, such as OPN and JAG1 in the BM of th3 mice. Interestingly, these molecules are directly regulated by PTH in OBs and MSCs [
49], and their reduction leads to a loss of HSCs quiescence [
50]. PTH is a key player of calcium and phosphate homeostasis, regulating bone remodeling and HSC maintenance via its specific receptor on BM stromal cells [
49]. Aprile and colleagues demonstrated that the downregulation of PTH- JAG1-Notch1 axis and OPN in BThal leads to defective stromal BM niche with impaired bone deposition and defective crosstalk between osteolineage cells and HSCs [
114]. These findings were also validated in patients’ samples.
Since MSCs also produce JAG1, the authors analyzed this population in th3 mice. MSCs are characterized by decreased frequency and lower expression of Jag1 as compared to wt controls [
114]. Consistently, MSCs from BThal patients showed reduced frequency and clonogenic potential, lower proliferation rate, impaired differentiation potential, and a reduced capacity to support HSPCs [
127].
SCD. Individuals suffering from SCD experience acute and chronic bone pain caused by occlusive events within the tissue vasculature that result in ischemia, necrosis, and organ degeneration [
34]. However, the pathophysiology of bone defects is still under investigated. Recent studies have suggested that environmental stimuli, such as inflammation, may influence the osteoporotic-like phenotype observed in SCD bone [
128,
129]. Because of the interactions between SS-RBCs and vascular endothelium, SCD patients display abnormally high concentrations of inflammatory molecules, especially IL-6, IL-8, and TNF-α. In healthy conditions, inflammation plays a crucial role in regulating bone remodeling; however, the chronic inflammation localized within the bone microcirculation may prolong OC activity via the upregulation of RANKL [
130,
131]. Micro-CT image analysis of transgenic SCD mice showed altered bone microarchitecture with fewer trabeculae and deteriorated structure, indicating progressive damage of SCD bone tissue [
132]. Dalle Carbonare et al. reported that recurrent hypoxia/reperfusion events, mimicking acute VOCs, activate osteoclastogenesis and bone turnover in SCD mice, with upregulation of the pro-resorptive cytokine IL-6 and suppression of osteogenic lineage markers, such as
Runx2 and
Sparc [
133]. The administration of zoledronic acid, a potent inhibitor of osteoclastogenesis and OC activity, ameliorated bone impairment and promoted osteogenic lineage. These data supported the view that bone disease in SCD is related to the biased coordination of remodeling signals towards bone absorption. In addition, the reduced OB recruitment and the increased OC activity are induced by local hypoxia, oxidative stress, and the release of IL-6. Furthermore, IO due to chronic transfusions was reported to increase bone resorption and impair the trabecular microarchitecture in SCD [
134].
Human MSCs derived from SCD BM were found to have altered expression of SCF and CXCL12, but showed normal in vitro functionality [
135]. However, recent work by Tang et al. reported a reduced frequency of MSCs in the BM of SCD mice, ROS accumulation, and a decreased adipogenic and osteogenic differentiation potential, also suggesting impaired MSC functional properties [
115]. Gene expression profiling revealed a decreased transcription of key niche molecules, such as
Opn, as well as vascular cell adhesion to protein 1 (
Vcam1)
, Angpt1,
Scf, and
Cxcl12, associated with an impaired ability to maintain HSCs in vitro and in vivo. These data are in line with increased HSC mobilization and reduced engraftment upon transplantation. Treatment with NAC and transfusions reduced MSC oxidative stress and improved the crosstalk between SCD MSCs and HSCs. Activation of TLR4 by hemolysis contributed to SCD MSC dysfunction.
Alterations in the BM vasculature were reported to be critical for SCD hematopoiesis too. Park et al. demonstrated that SCD mice have a disorganized BM vascular network with increased numbers of highly tortuous arterioles and fragmented sinusoidal vessels [
136]. In SCD, slow RBC flow and vaso-occlusions diminish local oxygen availability in the BM cavity and increase ROS production. Elevated levels of HIF-1α were found, triggering an enhanced neovascularization. Transplantation of BM cells from SCD mice into wt recipients recapitulated the SCD vascular phenotype by increasing HIF-1α signaling in normal mice. Conversely, blood transfusions completely reversed the altered vascular network, highlighting the plasticity of the BM vascular niche.
4. Hematopoietic and Soluble Niche Factors
BThal. Data on BThal mice indicate that multi-factorial alterations in the BM niche can impair HSC self-renewal and repopulating capacity. During the analysis of stromal components and soluble factors of the BM microenvironment, high systemic and BM local levels of FGF23 were detected [
137]. FGF23 is a negative regulator of bone metabolism and PTH secretion [
138], mainly produced by bone and erythroid cells in response to the anemia-related factor EPO [
139]. The enhanced activation of FGF23 signaling has been proposed as the mechanism underlying bone disease and low PTH levels in th3 mice, negatively impacting the functional crosstalk between HSCs and the stromal niche [
137].
In addition to the BM stroma, altered levels of multiple local and systemic factors were found, including SCF, ANGPT1, and CXCL12 [
114], as well as a reduction in serum TPO [
140]. Since TPO is a key regulator of both HSCs [
53,
141] and MKs [
73,
142], the TPO defect can have a dual role on BThal HSCs and BM microenvironments, thus contributing to the impaired HSC–niche crosstalk. Moreover, the condition of chronically reduced TPO stimulation in BThal is consistent with reported results of higher cycling activity of HSCs in the absence of TPO [
141], and effectively correlates with data of low HSC quiescence in th3 mice [
114]. Low TPO also impacts MK maturation and their downregulated expression of niche molecules in th3 mice [
140].
By focusing on other hematopoietic populations of the BM niche, different BM resident Mφs have been reported to indirectly regulate HSC retention by acting on niche stromal cells [
143]. Thus, the dissection of different populations of BM Mφs in BThal can contribute to the enhanced proliferation, increased mobilization, and reduced repopulating potential of th3 HSCs. Since BThal is characterized by IE with reduced erythroid terminal differentiation and expansion of the BM erythroid precursors, as expected, the frequency of erythroblastic island Mφs, essential for erythroblast survival and maturation, was significantly increased (unpublished data). BThal neutrophils were reported to display aberrant maturation and defective effector functions [
144]. Reduced BM Mφ–neutrophil interactions can play a role in BThal HSC mobilization, through the indirect effect on the production of CXCL12 retention molecule by the BM stromal niche [
82]. Preliminary data from the th3 mouse model showed an imbalanced polarization towards the M1 phenotype and a reduced neutrophil clearance by BThal BM Mφs, suggesting a potentially negative effect on HSCs (unpublished data).
SCD. In the complexity of SCD pathophysiology, many hematopoietic populations and soluble factors potentially involved in the regulation of the BM microenvironment homeostasis are altered [
145]. However, their direct contribution to the HSC niche is still completely unexplored.
Among the more studied populations, neutrophils of SCD patients and mice were activated by the increased production of ROS [
146]. They contribute to SCD pathogenesis by capturing circulating SS-RBCs, inducing VOCs, and secreting inflammatory cytokines [
147]. Free heme induces neutrophil extracellular trap (NET) formation by activated neutrophils, significantly contributing to SCD pathogenesis [
148]. In SCD mice, the aged neutrophil population is expanded and positively correlates with adhesion and interactions with RBCs. Neutrophil ageing is regulated by the microbiome [
149] and neutrophil clearance by BM was reported to modulate the HSC niche [
82]. Thus, the involvement of SCD neutrophils can be hypothesized in the regulation of the BM microenvironment.
Furthermore, platelets and monocytes in SCD were reported to have an activated phenotype with an active role in VOC pathogenesis, promoting the inflammation state associated with the disease [
150,
151]. SCD indeed have long been recognized as a chronic inflammatory disease, and, during infection or systemic inflammation, HSCs were reported to respond directly to inflammatory triggers [
152], leading to their activation, expansion, and enhanced myeloid differentiation [
153]. Furthermore, increased circulating heme and iron, i.e., hallmarks of SCD, were shown to induce Mφ phenotypic switching toward an M1 proinflammatory phenotype [
154], which has been reported to negatively regulate HSC maintenance [
81]. Whether BM cell populations and HSC function are altered in SCD in response to pro-inflammatory stimuli still needs to be explored.
5. The Role of IO in the β-Thalassemia Bone Marrow Niche
IO, associated with IE and therapeutic blood transfusions, is a key element of BThal pathophysiology. Despite improvements in chelation therapies over the past few years, the BThal BM niche accumulates a high content of iron. The direct impact of IO on BThal HSCs remains poorly characterized [
155]. Data on BThal th3 mice showed a positive enrichment of genes involved in iron homeostasis and significantly high levels of free reactive iron in HSCs, which correlate with increased ROS content (unpublished data).
Recently, Crippa et al. demonstrated that IO negatively affects BM MSCs in TDT patients [
127]. The in vitro exposure of BThal MSCs to increasing doses of iron revealed an upregulation of iron transporters, such as
ZIP14,
ZIP18, transferrin receptor 1 (
TFR1), and ferritin, thus suggesting that BThal MSCs can uptake and store iron. These findings are corroborated by the direct assessment of iron content in BThal MSCs using Perl’s staining. BThal MSCs display high ROS levels, as a result of the impaired antioxidant response, which correlates with a significant pauperization of the most primitive CD146
+ CD271
+ MSC pool. BThal MSCs showed reduced clonogenic capacity, lower proliferation rate, early cell cycle arrest, and impaired differentiation potential into adipocytes and bone. In addition, they express lower levels of hematopoietic supportive factors, such as SCF,
CXCL12, cadherin 2 (
CDH2),
VCAM,
ANGPT1, vascular endothelial growth factor A (
VEGFA),
IL-6, and
FGF2. Therefore, they fail to attract and expand HSCs in transwell migration assays and 2D co-culture experiments. Consistently, the in vivo transplantation of CD34
+ HSPCs, along with BThal MSCs, revealed a reduced hematopoietic engraftment upon xenotransplantation in NSG mice. Finally, the authors developed a humanized ossicle model, consisting of gelatin scaffold pre-seeded with MSCs, ECs, and CD34
+ HSPCs, to test the ability of BThal MSCs to form a proper BM niche in vivo. Transplantation of the humanized ossicle into NSG mice showed a delay in the formation of bone and vessels, as well as a reduced number of human CD45
+ hematopoietic cells ossicles derived from BThal MSCs.
Strikingly, treatment with the iron chelator deferoxamine (DFO) in the presence of iron decreased the expression of iron transporters, potentiated the antioxidant defense system in BThal MSCs, and rescued the expression of the hematopoietic supportive factors [
127]. Therefore, treatment with chelating agents or antioxidants can represent a therapeutic strategy to ameliorate BThal MSC supportive capacity, thus potentially improving the transplantation outcome.
Overall, these findings highlight previously unexplored multifactorial alterations of BM components in the biocomplexity of BThal and SCD (Table 1 and Figure 1). Elucidating the overriding players and the functional interconnections between stromal and hematopoietic alterations of the BM HSC niche will pave the way towards potential combined therapies, not only targeting the genetic defect, but also HSC and the BM microenvironment, in order to improve HSC transplantation and gene therapy approaches.
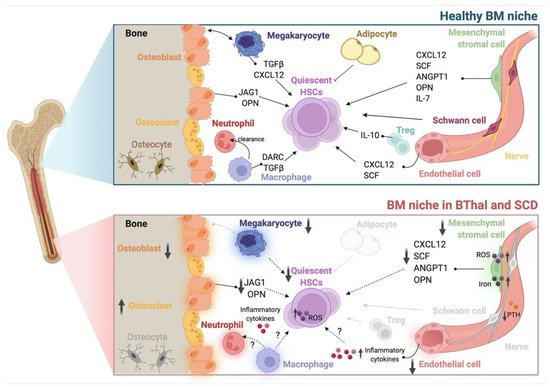
Figure 1. The BM HSC niche in healthy conditions and in BThal and SCD. Schematic representation of the adult BM niche in homeostasis, showing different stromal (osteoblasts, mesenchymal stromal cells, endothelial cells, Schwann cells, nerve fibers, and adipocytes) and hematopoietic (megakaryocytes, osteoclasts, macrophages, neutrophils, and Tregs) cell types and niche factors that regulate HSC quiescence and function (upper panel). The BM niche in BThal and SCD shows alterations in BM osteolineage cells, mesenchymal stromal cells, endothelial cells, and megakaryocytes, causing the reduced production of niche molecules supporting HSC activity. The accumulation of ROS, iron, and inflammatory cytokines contributes to the impairment of HSC maintenance (bottom panel). Created with BioRender.com.
Table 1. Altered BM niche populations in BThal and SCD. Alterations are shown as increased (⇑) or decreased (⇓) levels of specific features in BM niche populations.
| Cell Population |
Disease |
Species |
Alterations |
References |
| HSC |
BThal |
mouse |
⇓ number
⇓ quiescence
⇓ stemness
⇓ reconstitution capacity
⇑ response to stress |
[114] |
| human |
⇓ frequency
⇓ quiescence
⇓ stemness
⇑ response to stress (HSPC) |
[114,121,122] |
| SCD |
mouse |
⇓ frequency
⇑ ROS
⇑ DNA damage
⇓ quiescence (HSPC)
⇑ mobilization (HSPC) |
[115,123,124] |
| human |
⇓ frequency
⇑ mobilization |
[121,125] |
| Osteolineage cell |
BThal |
mouse |
⇓ BMD
⇓ systemic PTH
⇓ OB activity
⇓ niche molecules
⇑ FGF23 |
[114,126,137] |
| human |
⇓ niche molecules |
[114] |
| SCD |
mouse |
⇓ bone microarchitecture |
[132] |
⇑ osteoclastogenesis
⇓ osteogenic factors |
[133] |
| MSC |
BThal |
mouse |
⇓ frequency
⇓ niche molecules |
[114] |
| human |
⇓ frequency
⇓ osteogenic and adipogenic potential
⇑ ROS
⇑ iron content
⇓ niche molecules
⇓ HSPC maintenance |
[127] |
| SCD |
mouse |
⇓ frequency
⇑ ROS
⇓ osteogenic and adipogenic potential
⇓ niche molecules
⇓ HSC maintenance |
[115] |
| human |
⇓ niche molecules |
[135] |
| EC |
SCD |
mouse |
altered BM vasculature
⇑ inflammatory cytokines |
[136] |
| MK |
BThal |
mouse |
⇓ systemic TPO
⇓ maturation
⇓ niche molecules |
[140] |
| Neutrophil |
BThal |
mouse |
altered maturation |
[144] |
This entry is adapted from the peer-reviewed paper 10.3390/ph15050592