Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | George Kostakis | -- | 3201 | 2022-07-01 15:21:48 | | | |
| 2 | Camila Xu | -2 word(s) | 3199 | 2022-07-04 04:41:36 | | | | |
| 3 | Camila Xu | Meta information modification | 3199 | 2022-07-04 05:11:29 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Farhi, J.; Lykakis, I.N.; Kostakis, G.E. Recent Advances in A3 Coupling with Metal Salts. Encyclopedia. Available online: https://encyclopedia.pub/entry/24748 (accessed on 22 July 2026).
Farhi J, Lykakis IN, Kostakis GE. Recent Advances in A3 Coupling with Metal Salts. Encyclopedia. Available at: https://encyclopedia.pub/entry/24748. Accessed July 22, 2026.
Farhi, Jonathan, Ioannis N. Lykakis, George E. Kostakis. "Recent Advances in A3 Coupling with Metal Salts" Encyclopedia, https://encyclopedia.pub/entry/24748 (accessed July 22, 2026).
Farhi, J., Lykakis, I.N., & Kostakis, G.E. (2022, July 01). Recent Advances in A3 Coupling with Metal Salts. In Encyclopedia. https://encyclopedia.pub/entry/24748
Farhi, Jonathan, et al. "Recent Advances in A3 Coupling with Metal Salts." Encyclopedia. Web. 01 July, 2022.
Copy Citation
Recent advances in the metal salt catalysed multicomponent reaction of aldehydes, amines, and alkynes, known as A3 coupling, which yields propargylamines, a valuable organic scaffold.
A3 coupling
catalysis
multicomponent reaction
metal salts
copper
aldehyde
amines
alkynes
1. Introduction
Creating molecular complexity from readily available starting materials and catalysts is a long-standing and ongoing goal for synthetic chemists [1]. Synthetic methodologies that use more than two substrates and a metal-based catalyst, a subcategory of the well-known multicomponent reactions, involve complicated, possibly domino, organic transformations and yield valuable organic scaffolds in one step. These methodologies are inexpensive, produce minimal waste, and are atom- and energy-efficient, all significant advantages over traditional catalytic industrial protocols. Moreover, many such industrial protocols employ fossil fuels and well-characterised but expensive catalysts—considering the pressing need to transition to “green” processes, a modular and selective polycomponent reaction starting from readily available material is highly desirable.
One multicomponent reaction is the A3 coupling (Scheme 1), named for its three components: an aldehyde, an alkyne and an amine. This versatile reaction constitutes the most straightforward paradigm for a multicomponent reaction. It combines two unique catalytic concepts: C-H activation of the alkyne moiety and use or in situ formation of imines or enamines. Furthermore, the ideally chiral propargylamines (PA) formed by this reaction are essential precursors in organic synthesis for various products, including isoindolines [2], oxazolidines [3], pyridines [4][5], and alkaloids [6][7][8] (Scheme 2). Due to this importance, it is necessary to understand the catalyst’s role and develop protocols to yield the final product in high enantiomeric excess.
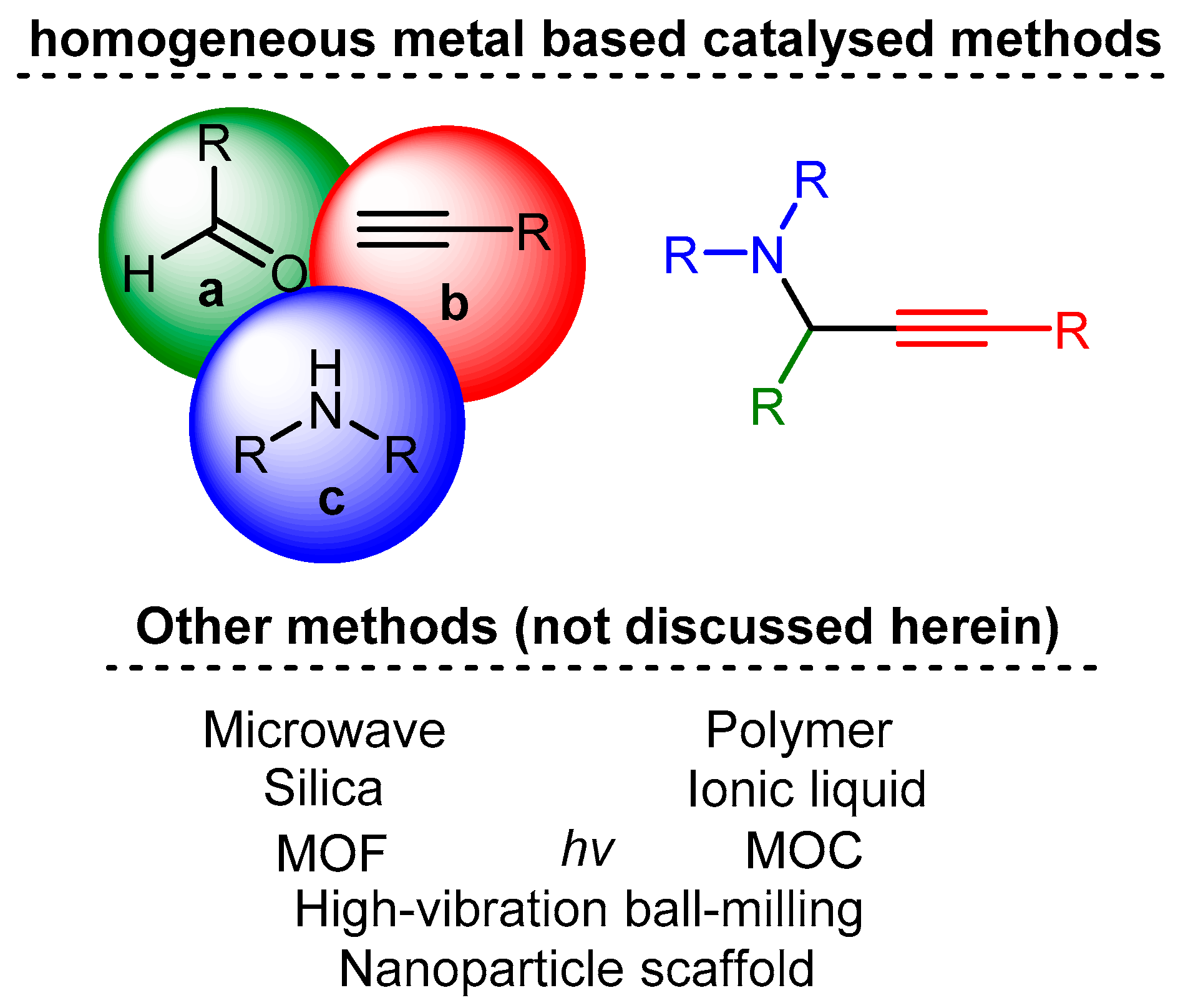 Scheme 1. The A3 coupling reaction.
Scheme 1. The A3 coupling reaction.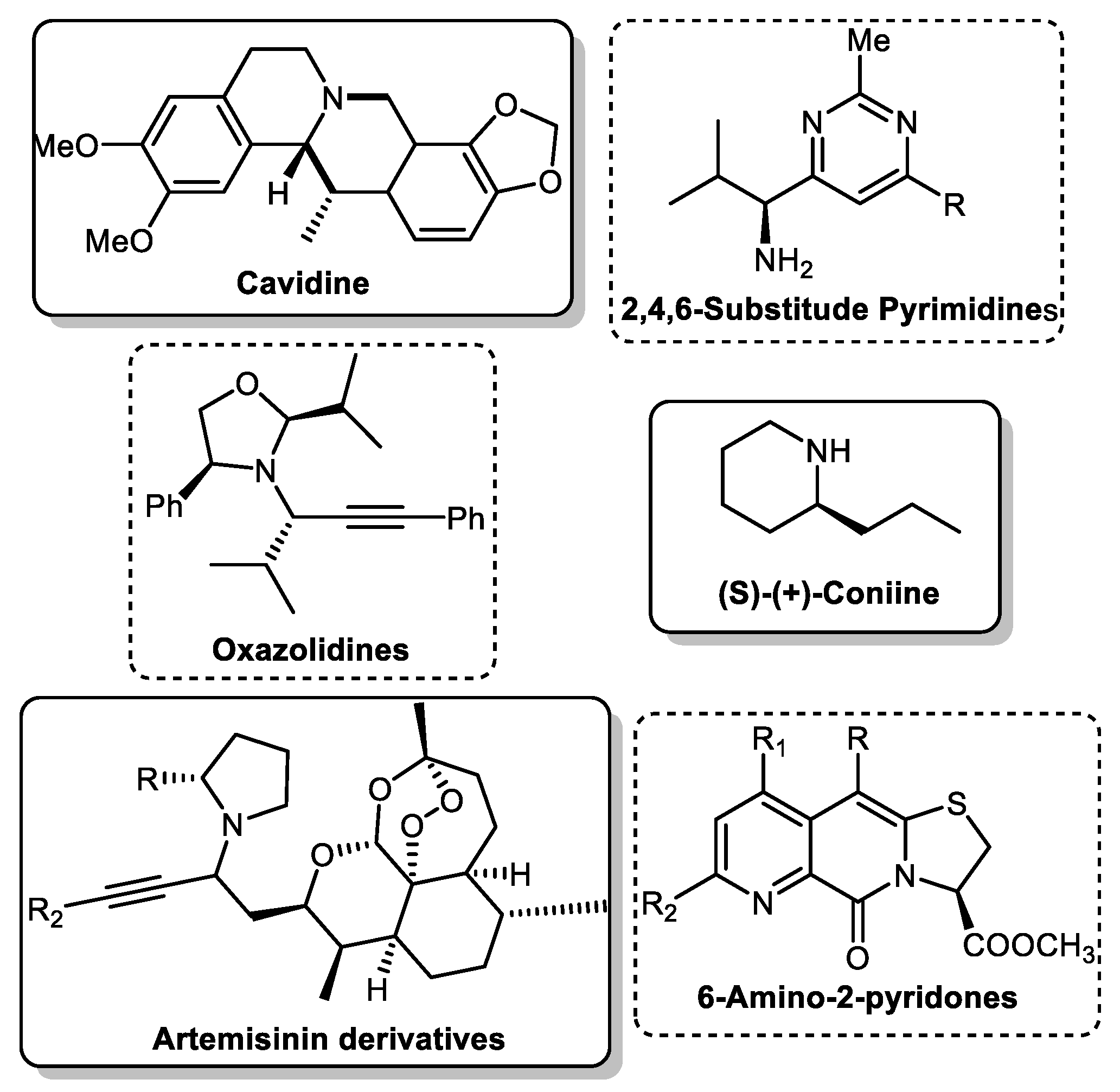 Scheme 2. Natural and building blocks synthesised from PAs.
Scheme 2. Natural and building blocks synthesised from PAs.The C-H activation has gained significant interest and represents one of the eighth main research milestones [9][10][11]. Similarly, imines and enamines are essential components for several organic transformations and organic scaffolds of high interest [12][13][14][15][16][17]. The A3 coupling reaction forms a new C-C σ bond via C-H activation of a terminal alkyne and yields PAs in one step with one water molecule as a by-product. Late transition metal catalysts effectively catalyse the A3 reaction. Mechanistic studies have identified the in situ formation of a π complex between the metal salt and the triple bond of the alkyne component. This intermediate partly shifts electron density from the alkyne to the metal cation, weakening the C-H sp bond, thus allowing proton removal by a weak base. The deprotonation of the π complex results in σ activation and formation of the acetylide, which can be subsequently added to the imine [18][19]. Initial reports on the A3 coupling reaction demonstrated that the reaction could be catalysed by silver [20], gold [21], and copper [22] via a one-pot synthesis. Other studies demonstrated various metals, including cadmium [23], cobalt [24], iron [25], mercury [26], indium [27], manganese [23], nickel [28], tin [23], zirconium [29], and zinc [30][31][32][33][34] facilitate the A3 coupling. These reactions often require an inert atmosphere, high temperatures, and long reaction times. Moreover, while several plausible reaction mechanisms have been proposed, in situ conditions make it difficult to analyse the catalytic mechanism effectively: for example, some hypotheses assume a monomeric active species [35][36][37][38][39], while others claim the active species is a paddlewheel dimer that would make the formation of a catalytic monomer difficult [8][40]. In contrast, other studies have suggested the existence of a dicopper catalyst instead of the monomeric species [41]. A more comprehensive understanding of the A3 catalytic mechanism would facilitate asymmetric synthesis developments and advance other reactions involving alkyne activation via a metal catalyst.
Notably, PAs can be formed at elevated temperatures without a metal catalyst [42], whilst various other methodologies successfully yield PAs. Protocols that involve polymers [43][44][45], silica [46][47], metal-organic cages [48], metal-organic frameworks (MOFs) [49][50], high-vibration ball-milling [51], photoredox [52], ionic liquids [53], microwaves [36], and nanoparticles [54][55] have been reported. Recent studies have developed a library of organic ligands that can bind in situ with a metal salt to produce various PAs in high yields and enantiomeric excess. While the scope of these reactions may be limited to a specific subset of components such as aromatic amines, a clear trend has emerged regarding useful metal salts and ligands: remarkably, a copper salt and an atropoisomer with a high barrier to rotation [56]. These atropoisomeric co-catalysts often have several non-covalent stabilising interactions that activate the imine and metal acetylide, including π-stacking rings [8][40][56][57][58][59], hydrogen bonding [60], and a combination of hard-soft interactions [56][60].
2. A3 Coupling with Metal Salts
The initial investigations of the transition-metal catalysed A3 coupling reaction to form PAs involved common metal salts such as copper, silver, and gold, with copper being the metal of choice due to its low cost.
2.1. Copper Salts
The initial investigations of the A3 coupling reaction incorporated Cu(I) salts. In an initial study by Li [22], a combination of two different metal sources, CuBr and RuCl3, was used to add phenylacetylene to an imine formed from aniline and aromatic or aliphatic aldehyde derivatives (Scheme 3). Notably, the reaction involved acetylide addition to an imine, a more challenging substrate than the more electronegative iminium made via secondary amines. To the best of our knowledge, this work remains one of the most efficient A3 coupling protocols involving aniline.
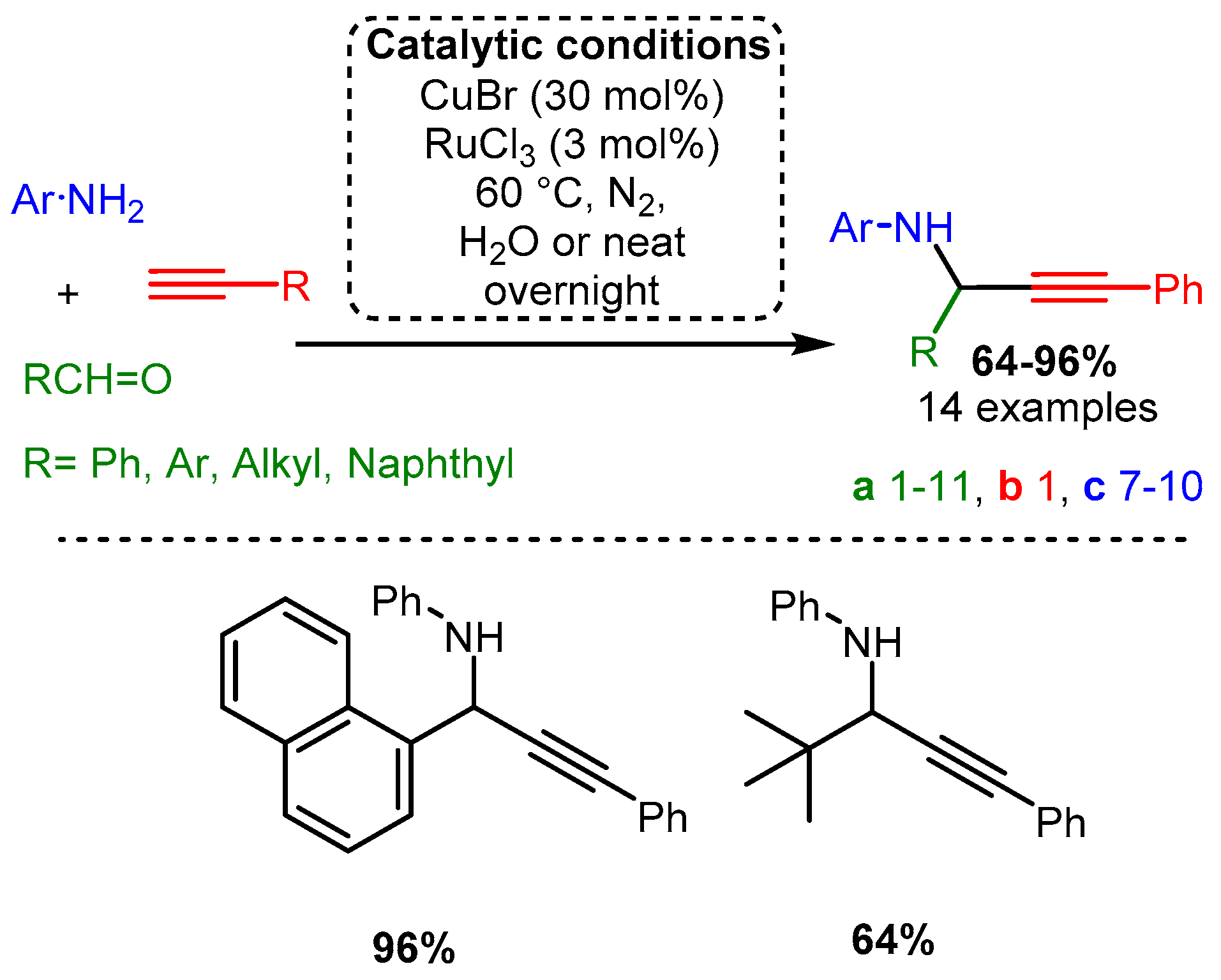 Scheme 3. (Upper): catalytic conditions and representative PAs with a range of yields and number of entries. (Lower): highest and lowest yielding PA. Coloured entries corresponding to the starting materials are provided whenever possible to facilitate correlation with the provided database. The following Schemes have a similar structure, and explanations will be provided if necessary.
Scheme 3. (Upper): catalytic conditions and representative PAs with a range of yields and number of entries. (Lower): highest and lowest yielding PA. Coloured entries corresponding to the starting materials are provided whenever possible to facilitate correlation with the provided database. The following Schemes have a similar structure, and explanations will be provided if necessary.Similarly, Iqbal and co-workers reported a procedure using solely CuCl to synthesise quinoline derivatives [61]. In the reported method, an aniline or other substituted arylamines (e.g., 4-methoxy aniline), aromatic aldehyde, and alkyne were sequentially added and reacted at 70 °C for up to 10 h, yielding a PA intermediate that subsequently cyclised into the functionalised quinoline product (Scheme 4). The proposed in situ generated Cu-acetylide intermediate reacts with the imine leading to the formation of the corresponding PA. After a propargyl–allenyl isomerisation cyclisation process, the desired quinoline derivative was formed under copper-catalysed conditions (mechanism not shown).
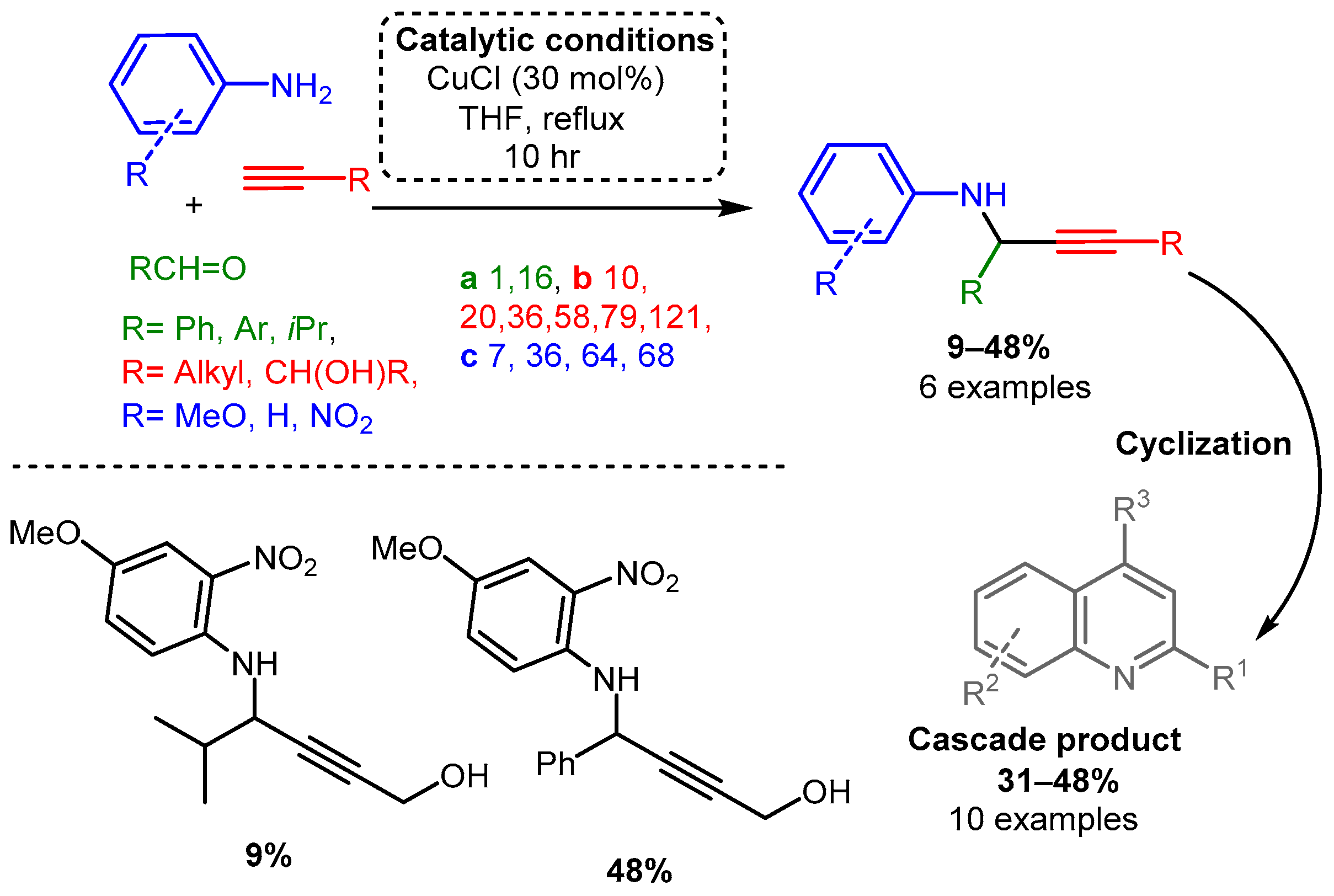 Scheme 4. Cyclisation cascade to form quinoline derivatives via PA intermediates.
Scheme 4. Cyclisation cascade to form quinoline derivatives via PA intermediates.In the same year, CuBr was used to catalyse the addition of alkynes to enamines, producing Pas [40]. Various alkynes were used while the enamine substrate was kept relatively constant: secondary amine (dibenzylamine, diallylamine) and aliphatic aldehyde. Researchers proposed that the enamine tautomerises to an iminium, which subsequently reacts with the activated alkyne (Scheme 5). It is worth noting that in the presence of R-(+)-Quinap (5.5 mol%) in toluene and at room temperature within 24 h, the corresponding enantiomeric PAs were synthesised in high yields and good to high enantiomeric excess, % ee (Scheme 5), these results will be discussed in detail in the related chapter.
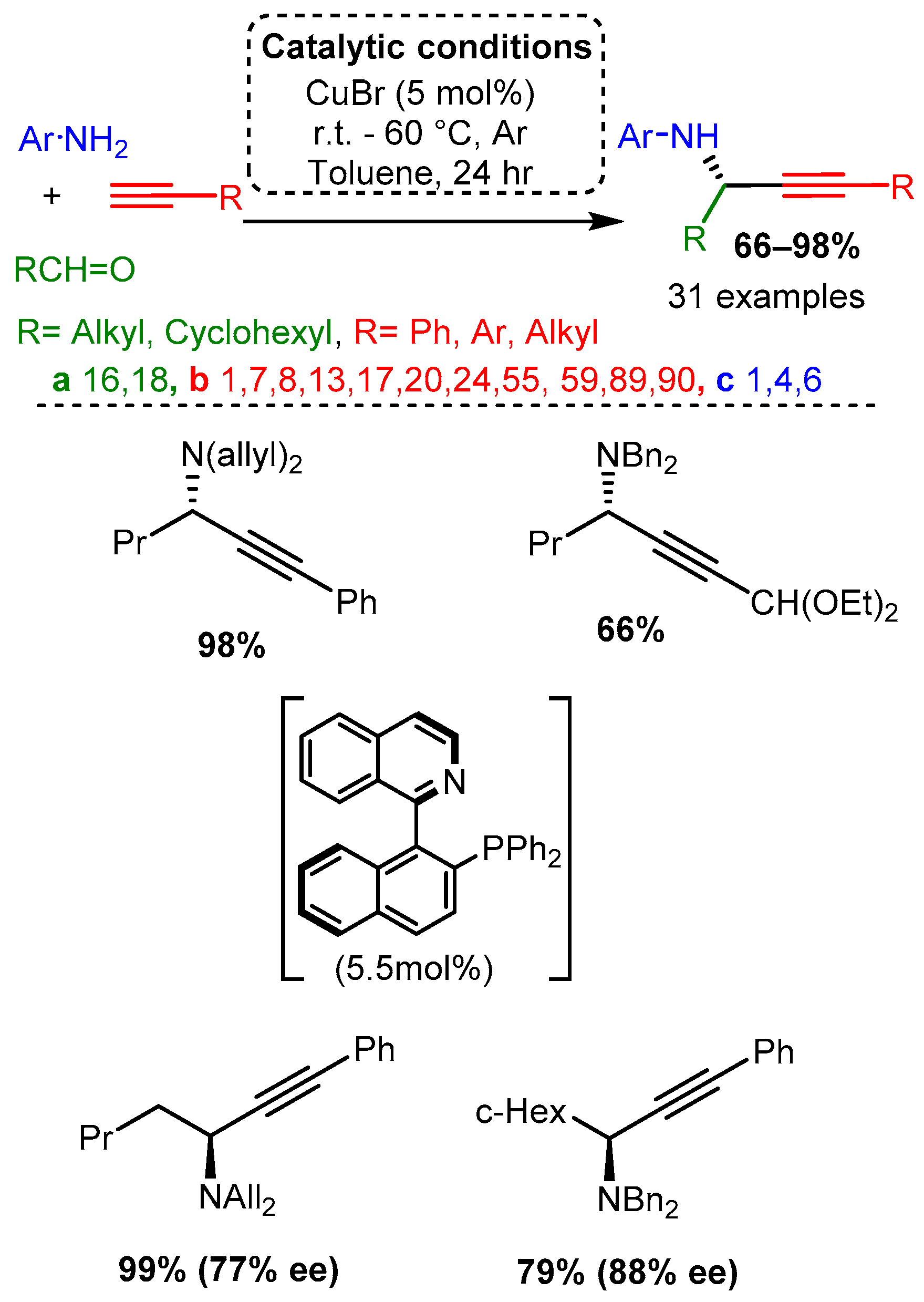 Scheme 5. PA access from premade enamines and synthesis of optically pure enantiomers.
Scheme 5. PA access from premade enamines and synthesis of optically pure enantiomers.The use of scaffolds containing secondary and tertiary amines were exploited to diastereoselectively synthesise PAs using a CuBr catalyst without an atropoisomer or other ligand [62]. High yields were achieved with aliphatic and aromatic alkynes and aldehydes. The proposed mechanism involved a diamine component complexing with the CuBr dimer to form a restrictive catalytic site, similar to the ligands discussed later in this research. In a later step, these PAs were converted to allenes using a CuI catalyst. Of note, the same substrates produced PAs with high yields and diasteromeric ratio (dr) when ZnI was incorporated under reflux conditions (Scheme 6).
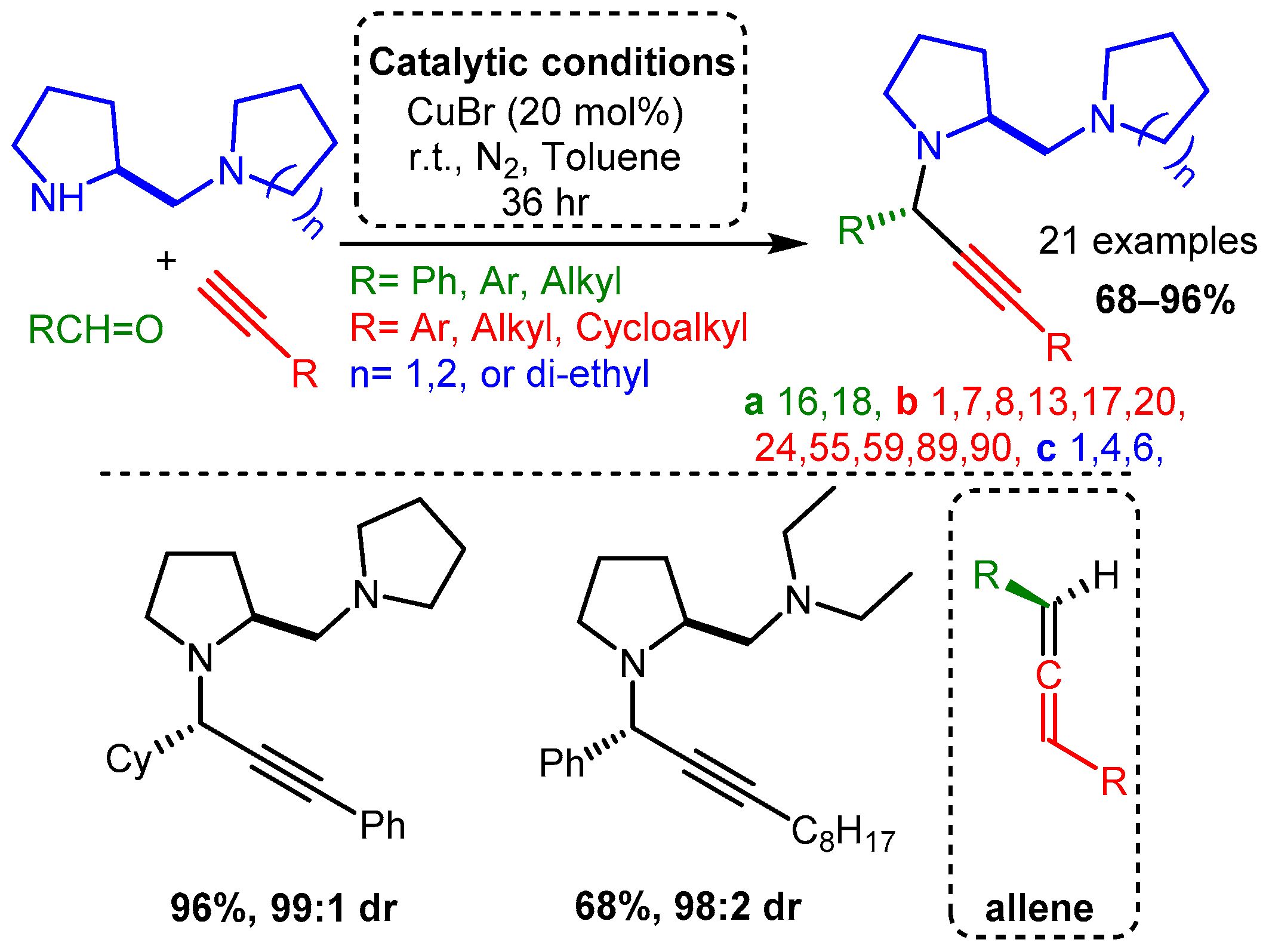 Scheme 6. Diastereoselective control of PA product using diamine substrates Post catalysis, the PAs could be converted to the corresponding allenes (bottom right).
Scheme 6. Diastereoselective control of PA product using diamine substrates Post catalysis, the PAs could be converted to the corresponding allenes (bottom right).The unique structural features of cyclic PAs incorporating scaffolds with two terminal amines and formaldehyde were exploited using a microwave-assisted CuCl catalyst in 1,4-dioxane to form functionalised, N-containing heterocycles [63]. While the substrates for this reaction were restricted due to the cyclisation cascade, the produced substituted heterocycles demonstrate the applicability of the A3 coupling reaction, especially when considering the chemoselectivity of the unsymmetrical dialkynylation (Scheme 7).
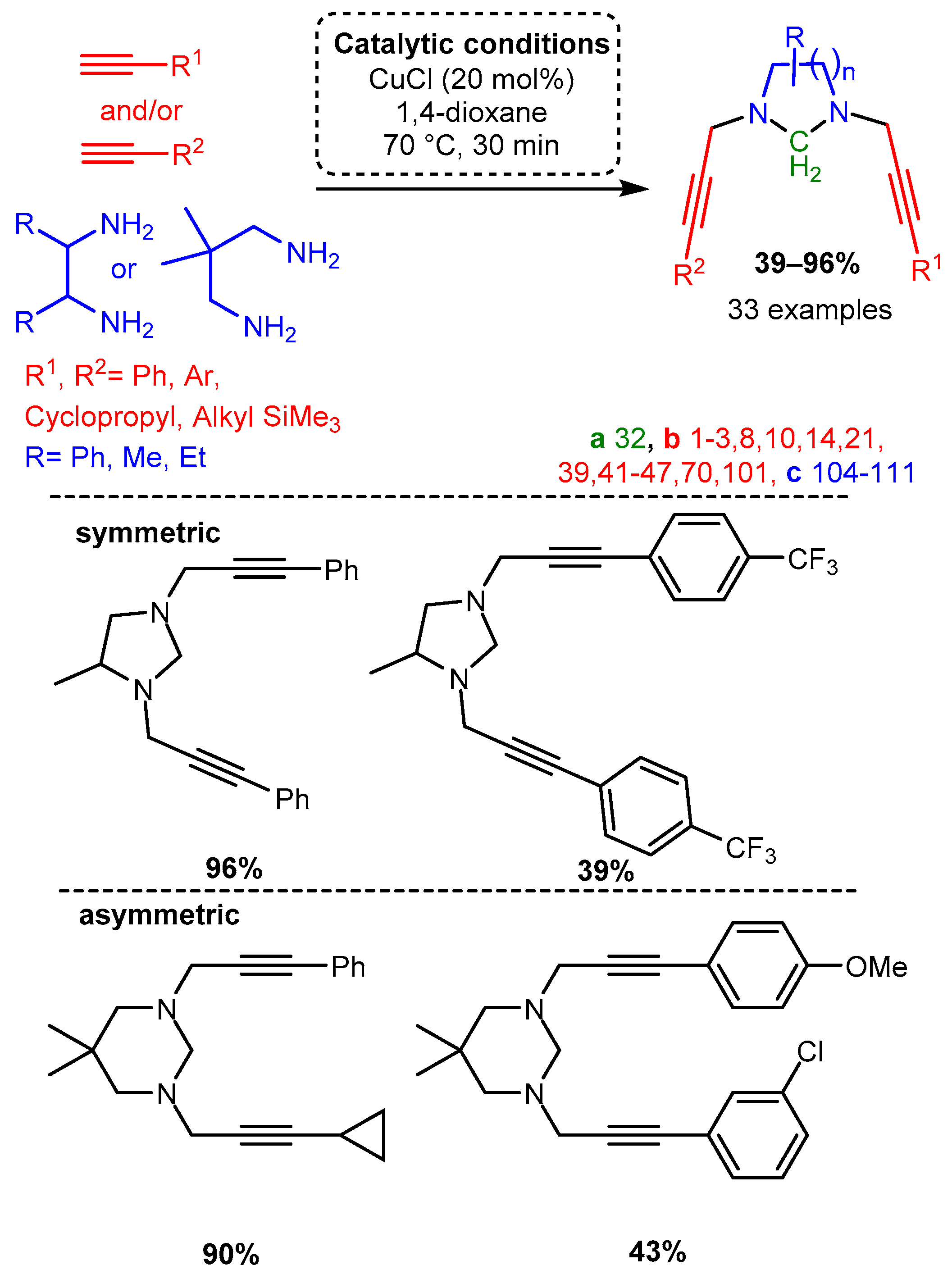 Scheme 7. Formation of cyclic divalent PAs from diamines with the appropriate component choice. Chemoselective alkynylation forms unsymmetrical products.
Scheme 7. Formation of cyclic divalent PAs from diamines with the appropriate component choice. Chemoselective alkynylation forms unsymmetrical products.Similarly, the combination of two different Cu(II) salts and amino alcohol as the amine component facilitated a cascade reaction that led to the formation of chiral oxazolidines [64]. In the optimised reaction, the combination of CuBr2 and CuCl2, each 10% loading, with the presence of the chiral plenylglycinol as the precursor and in solvent-free conditions, resulted in the target products in good yields (up to 90%) and excellent diastereomeric ratio (dr > 20/1) with a wide range of substrates (Scheme 8).
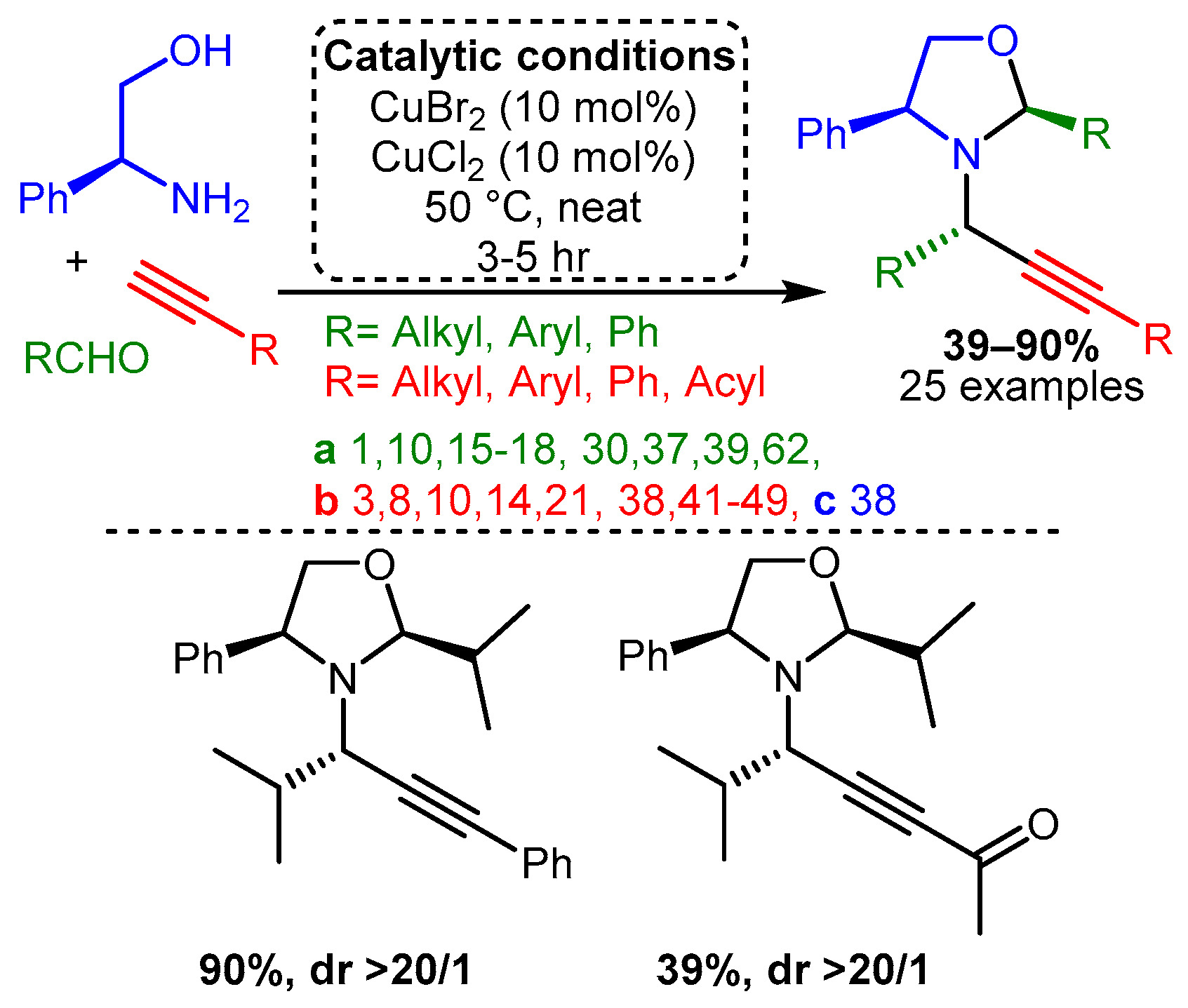 Scheme 8. Formation of monocyclic oxazolidines from chiral amino alcohols.
Scheme 8. Formation of monocyclic oxazolidines from chiral amino alcohols.Dos Santos and co-workers demonstrated that different alkynols could be used as a substrate with a CuCl catalyst to produce the corresponding hydroxy-PAs. The resulting hydroxy-PAs were achieved in moderate to high yields and could be converted in subsequent steps to alkaloids under a simple reduction process and intramolecular cyclisation [6] (Scheme 9).
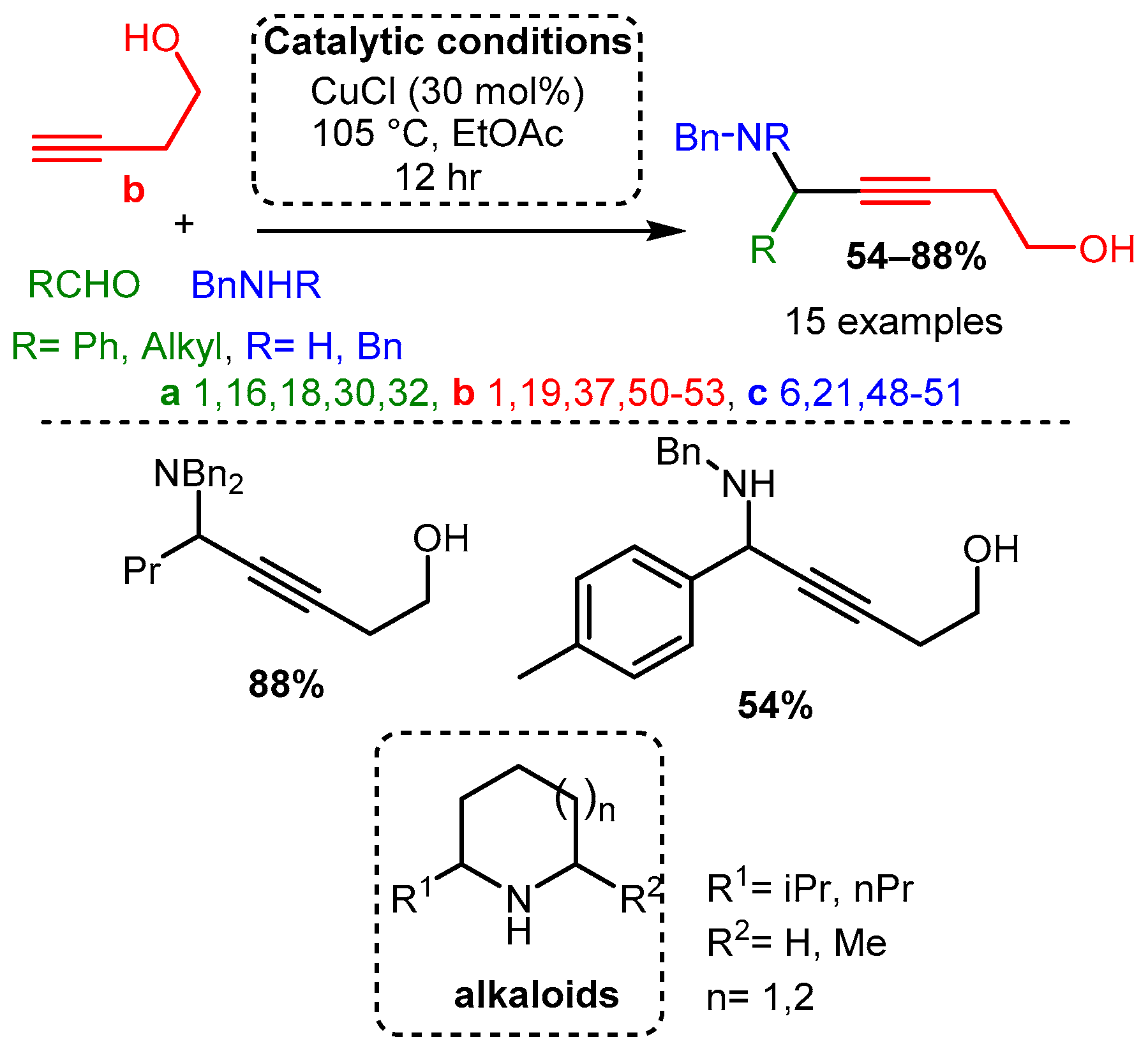 Scheme 9. A3 coupling with alkynols and further formation of alkaloids after a reductive/cyclisation cascade reaction.
Scheme 9. A3 coupling with alkynols and further formation of alkaloids after a reductive/cyclisation cascade reaction.Similarly, the use of propynal derivatives with a CuI catalyst yields 3-amino-1,4-diynes [65]. Interestingly, when subjected to the A3 coupling (propynal, amine, alkyne), asymmetric 3-amino-1,4-diynes were accessed in moderate to high yields (Scheme 10).
 Scheme 10. The use of propargyl derivatives allows access to symmetric and asymmetric 3-amino-1,4-diynes.
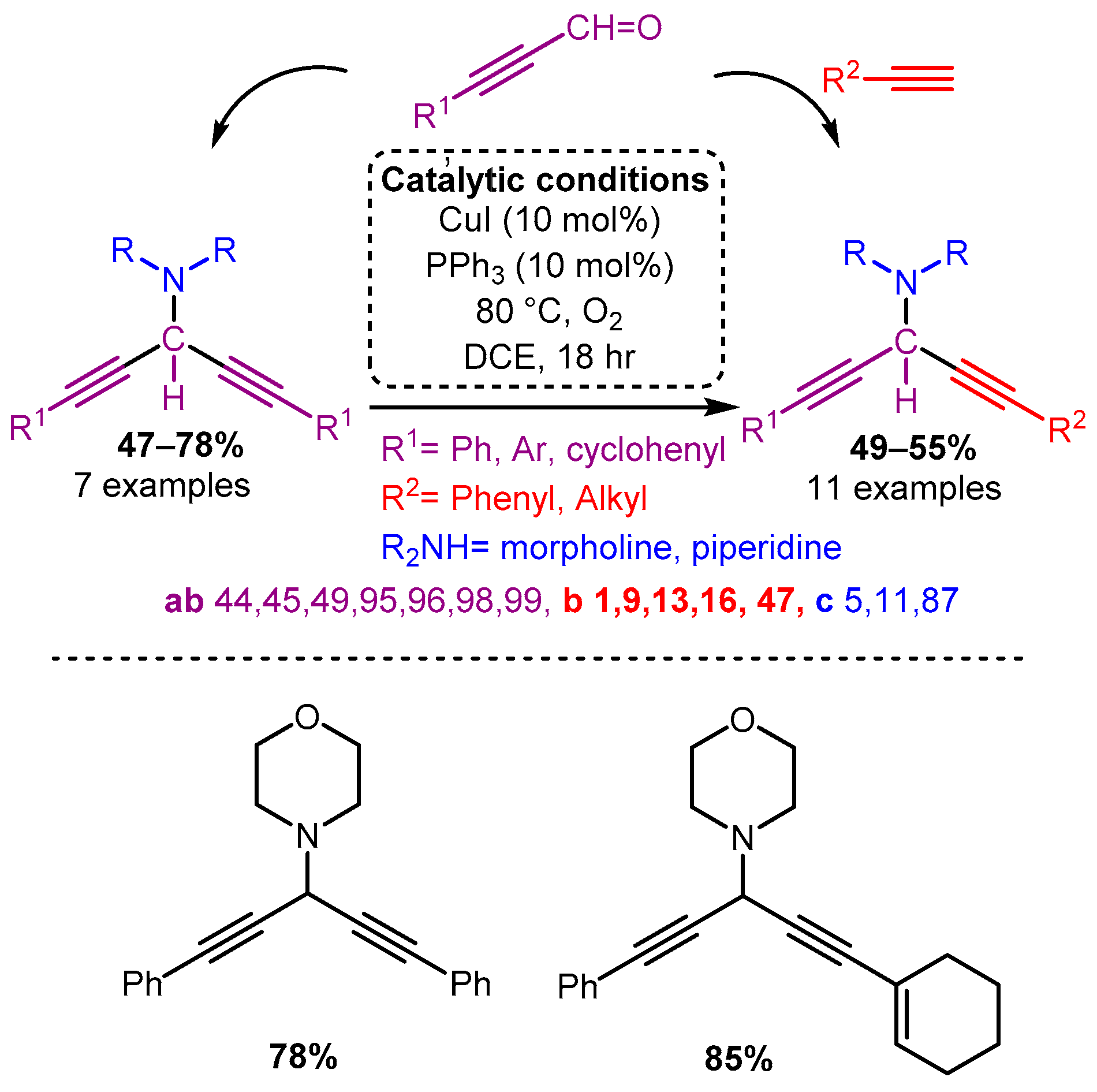
Scheme 10. The use of propargyl derivatives allows access to symmetric and asymmetric 3-amino-1,4-diynes.Using propiolic acid and its derivatives, PAs can be formed following a decarboxylative coupling path. This variation of the A3 coupling could be achieved at elevated temperatures without a metal-based catalyst but only when formaldehyde was incorporated as the aldehyde component [42] and with various aromatic and aliphatic aldehydes when CuI is involved as a catalyst [66] (Scheme 11 and Scheme 12).
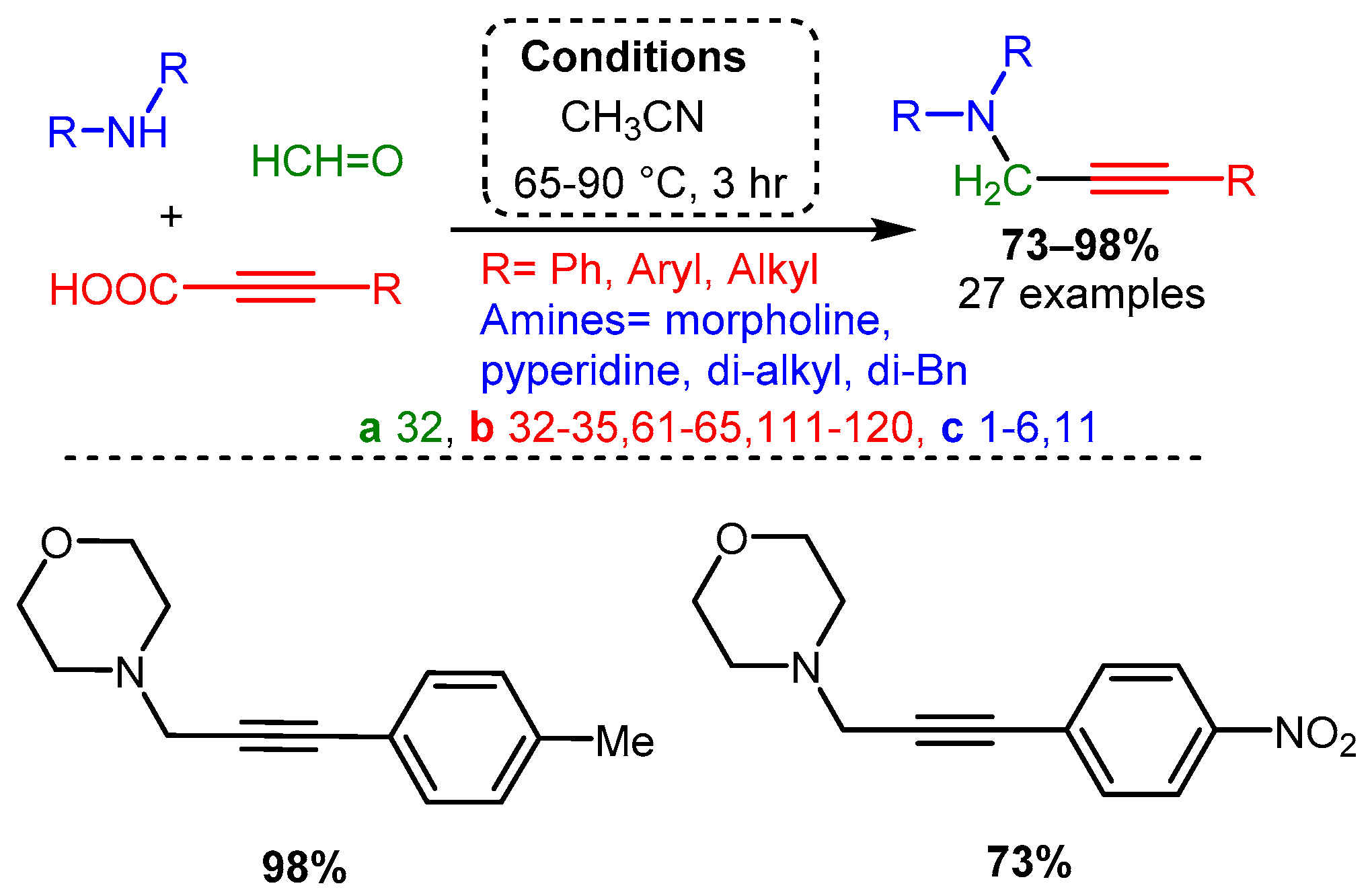 Scheme 11. The A3 decarboxylative coupling without catalyst.
Scheme 11. The A3 decarboxylative coupling without catalyst.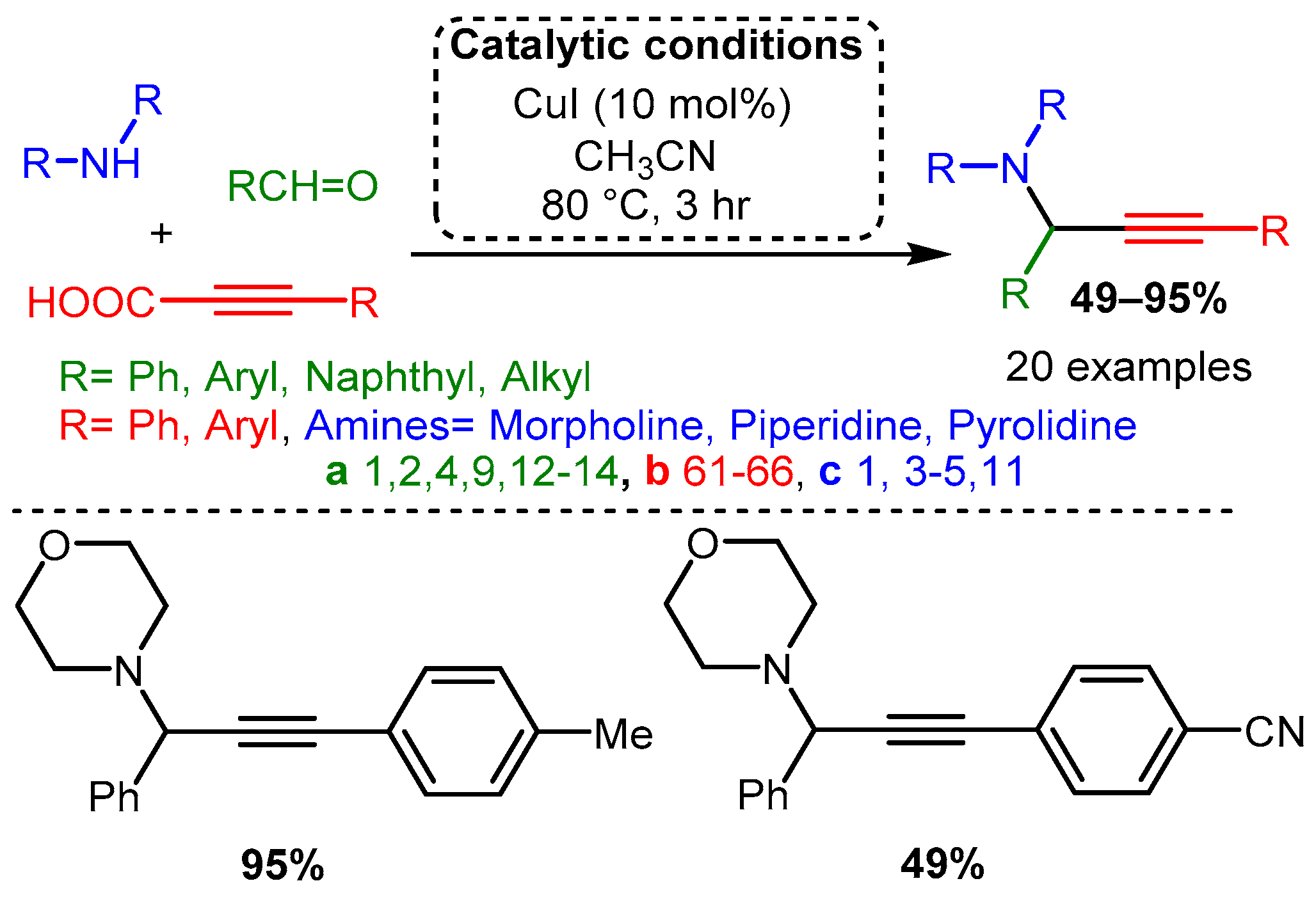 Scheme 12. The A3 decarboxylative coupling in the presence of CuI.
Scheme 12. The A3 decarboxylative coupling in the presence of CuI.The apparent advantage of this method is the increased stability of the alkynyl carboxylic acids. However, the applicability of this offshoot of the A3 coupling still needs to be explored.
The Cu(I) salts presented thus far have catalysed the A3 coupling reaction with various substrates. High yields have been achieved with more challenging substrates, such as primary amines and aliphatic alkynes, and creative choice of substrates has even allowed access to highly diastereoselective reactions. Nevertheless, many of these experiments are limited in scope; for example, the copper/ruthenium catalytic system presented by Li, while notable for its use of aniline derivates, limited the other substrates to phenylacetylene and aldehydes without α-hydrogens [64].
Many more recent studies have shifted attention to Cu(II) salts due to their ease of handling and lower costs, although their use without a chiral ligand has not been extensively explored. Larsen reported a Cu(OTf)2 catalyst for the A3 coupling of the electron-deficient (tosylated) nitrogen sources with alkyl, aryl, and heteroaryl aldehydes. This catalyst was able to catalyse the A3 variant, the ketone–alkyne–amine coupling reaction (KA2), which substitutes a ketone for an aldehyde [67] (Scheme 13). While initial screenings used a pre-formed imine component, it was later found that the three-component, one-pot reaction provided a higher yield (63% vs. 79%) and at a rate 20 times faster (results not shown). This observation suggests that the reaction proceeds via imine protonation to form an iminium intermediate instead of imine-copper coordination. To gain insights into the identity and function of the catalyst, a mixed catalyst study was performed. The addition of Cu(I) salts, the assumed catalytic species, did not increase the rate while doubling the concentration of Cu(OTf)2 doubled the reaction rate. These results cast doubt on the assumed Cu(I) active species and highlight the importance of the counterion in affecting the chemical environment to allow for effective catalysis.
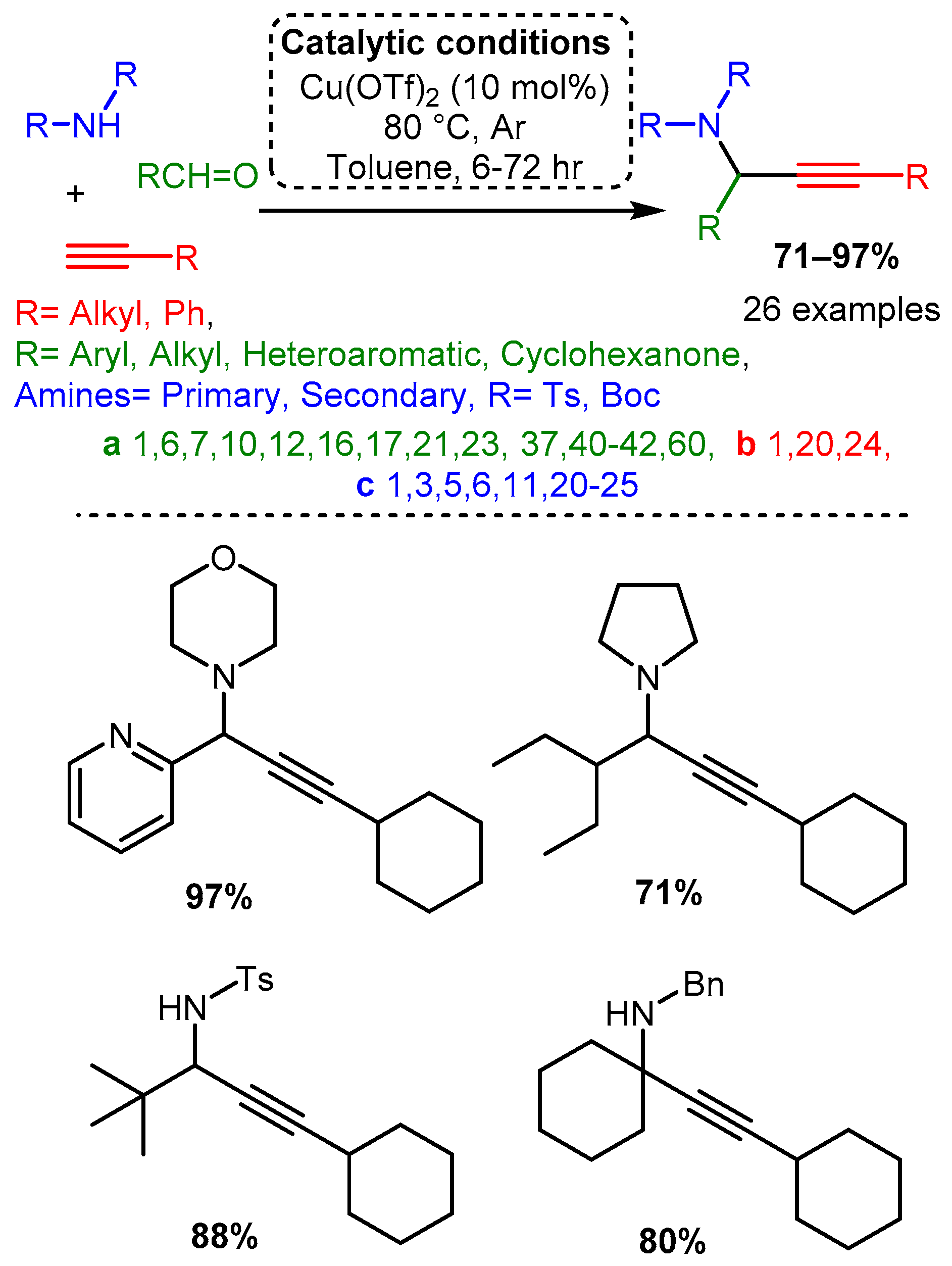 Scheme 13. Larsen et al. [67] used a Cu catalyst to catalyse the A3 coupling reaction with electron-deficient amines.
Scheme 13. Larsen et al. [67] used a Cu catalyst to catalyse the A3 coupling reaction with electron-deficient amines.Delpiccolo and co-workers employed a variety of substrates using a Cu(OTf)2 catalyst [68]. An aliphatic alkyne and cyclic secondary amine were used with differently substituted aromatic aldehydes. These data indicate that a small substituent at the ortho or para position increases yield compared to an unsubstituted aromatic aldehyde, while a substituent at the meta position decreases yield. Moreover, adding a nitro group to the aromatic aldehyde led to a significant drop in yield, while 4-methoxybenzladehyde and 4-fluorobenzaldehyde had approximately equal yields. Substitution of halides at the para position resulted in interesting trends: 4-chlorobenzaldehyde led to a precipitous drop in yield, while 4-bromobenzaldehyde restored high yield but resulted in an allene side product (Scheme 14). The synthesis of PAs in the solid phase was also reported, starting with the immobilised 4-formylbenzoic acid by anchoring to Wang resin. The synthesis of five different p-HOOC- or p-MeOOC-substituted PAs was achieved in media to good isolated yields, from 10% to 65% (Scheme 14).
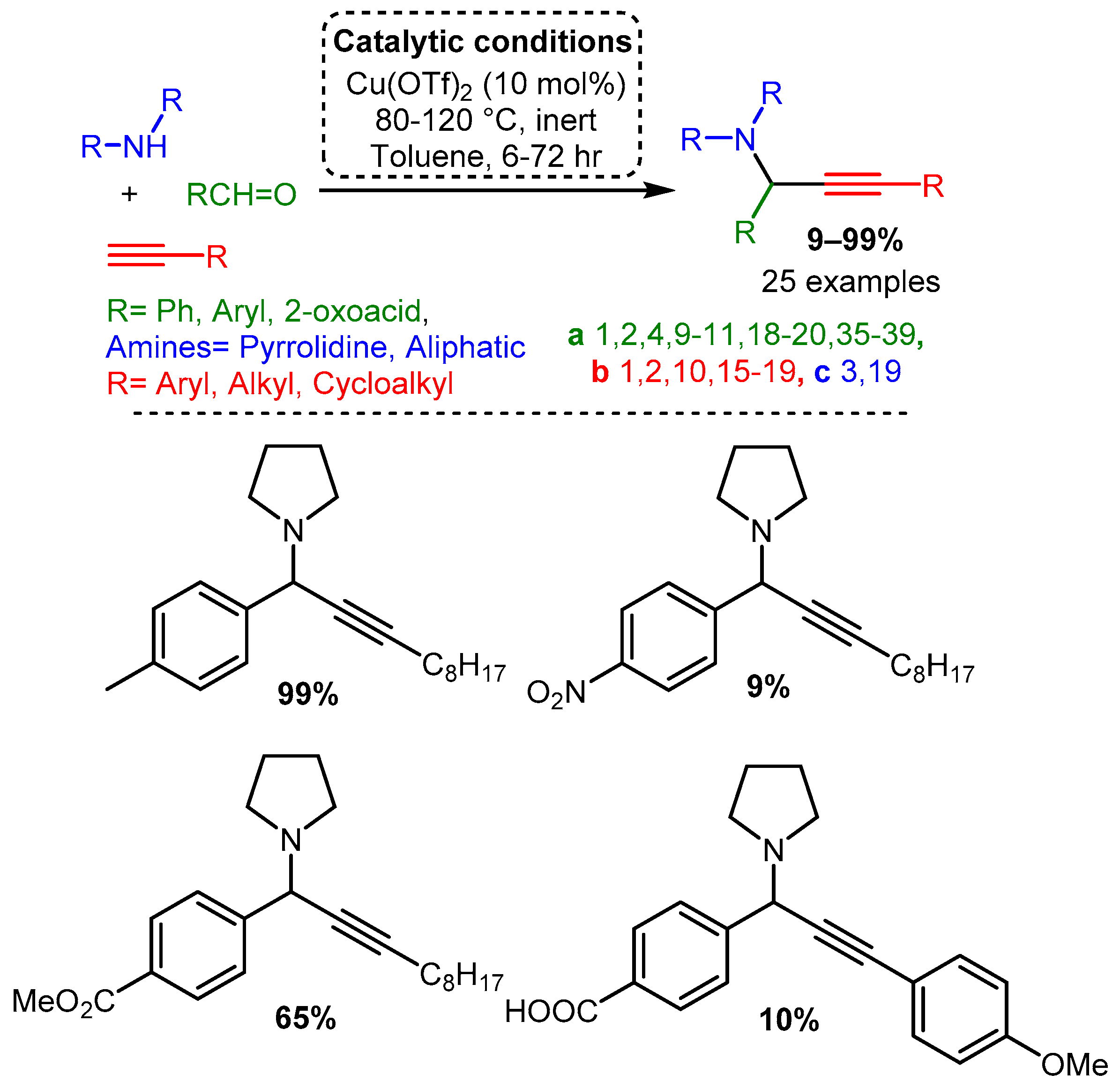 Scheme 14. Synthesis of PAs through an A3 coupling in homogeneous and solid-phase conditions.
Scheme 14. Synthesis of PAs through an A3 coupling in homogeneous and solid-phase conditions.2.2. Silver and Gold Salts
Initial studies of the A3 coupling reaction also investigated the efficacy of silver and gold salts as catalysts [20][21]. The first silver-catalysed A3 coupling reaction was reported by Li in 2003 [20] and the optimised catalytic protocol incorporated water, as the solvent, at 100 °C with 1.5–3 mol% loading of AgI. The PAs were synthesised in moderate to high yields, with aliphatic aldehyde components being the highest-yielding substrate. In contrast, aromatic aldehydes required prolonged times and produced PAs in lower yields. Moreover, only trace amounts of product were obtained when acyclic amines were used (Scheme 15).
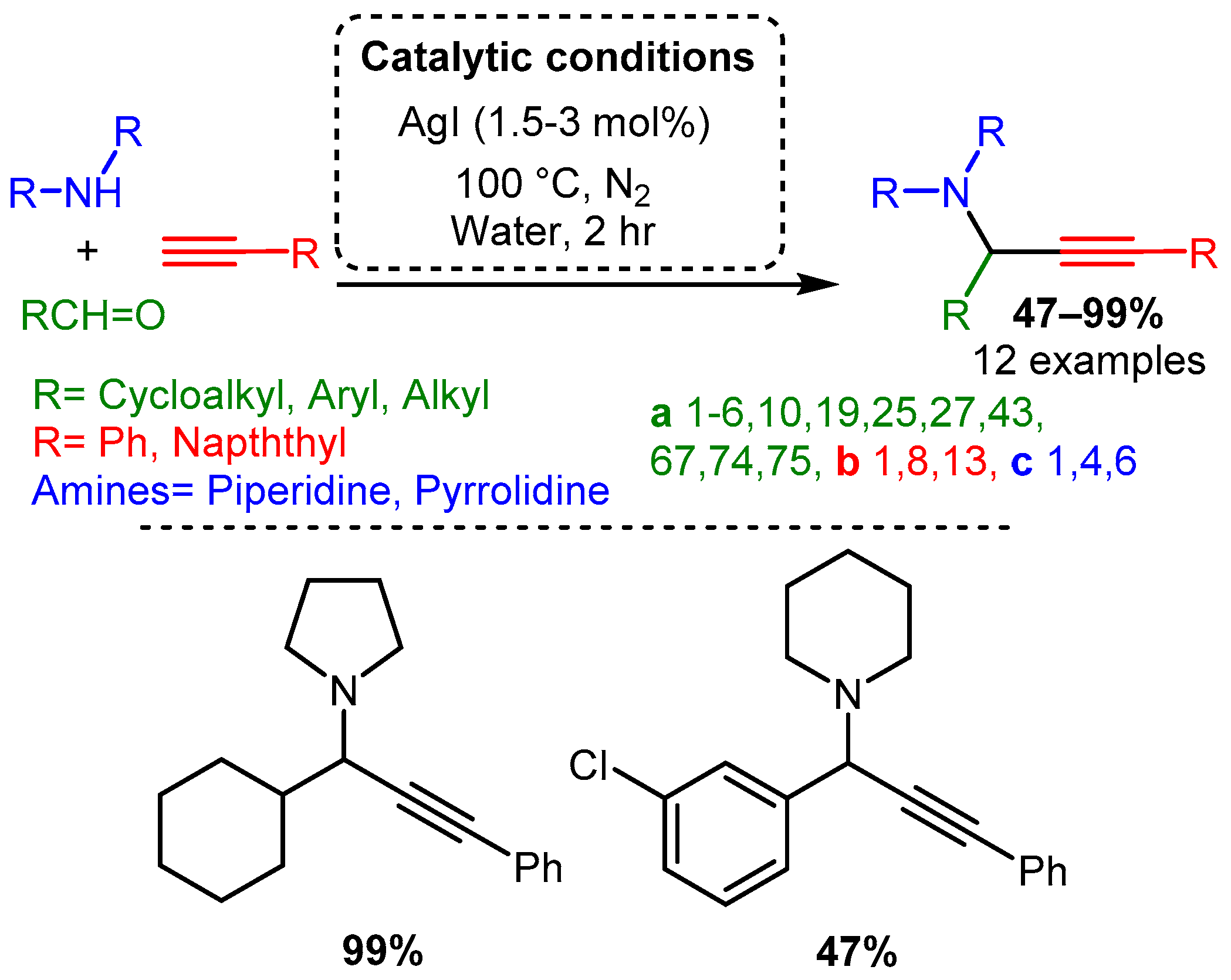 Scheme 15. Optimised conditions for Ag(I) salts.
Scheme 15. Optimised conditions for Ag(I) salts.Early work by Li [21] demonstrated that Au(I) and Au(III) salts (AuCl, AuI, AuBr3, and AuCl3) could be used to catalyse the reaction with a variety of substrates. The optimised reaction used an AuBr3 catalyst under an inert atmosphere in water. The reaction produced high yields even with 0.25% catalyst loading. Moreover, unlike the silver or copper salts screened by Li, the gold catalyst worked well with aliphatic and aromatic aldehydes (Scheme 16).
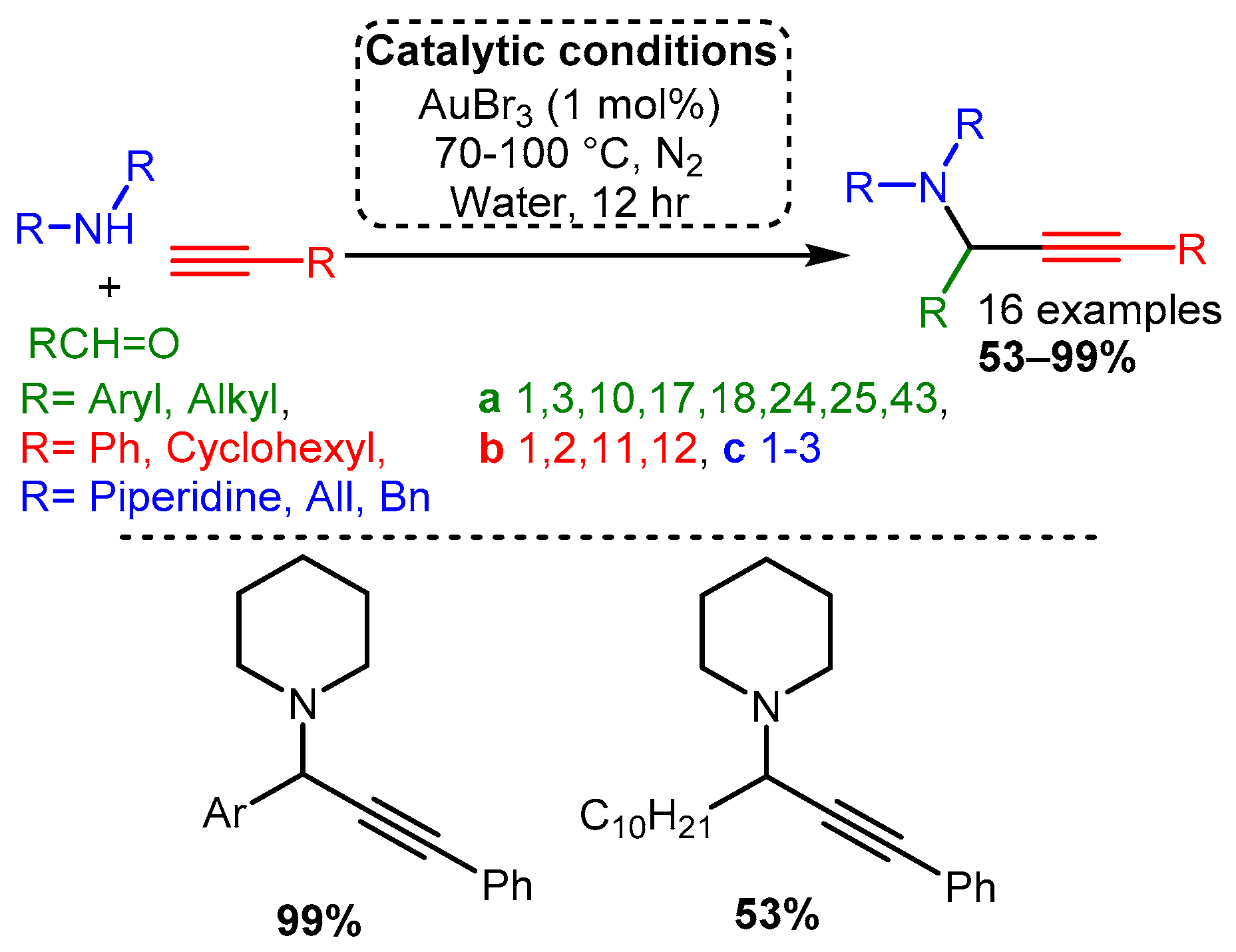 Scheme 16. Optimised conditions for Au(III) salts.
Scheme 16. Optimised conditions for Au(III) salts.According to the extensive classification, gold was the only group XI catalyst able to catalyse the A3 coupling reaction when α-oxyaldehydes were used as substrate [69]. Using higher catalytic loadings (5 mol%), the reaction at room temperature under an inert atmosphere produced good yields with a moderate preference for one diastereomer. This diastereoselectivity most likely resulted from the coordination of the iminium intermediate, thereby biasing acetylide addition (Scheme 17). Interestingly, while Ag(I) catalysts were unable to catalyse the A3 coupling reaction when an α-hydroxyaldehyde was used as a substrate, they were more effective than gold catalysts when α-alkylaldehydes were used. However, this variation was not extensively studied (results not shown).
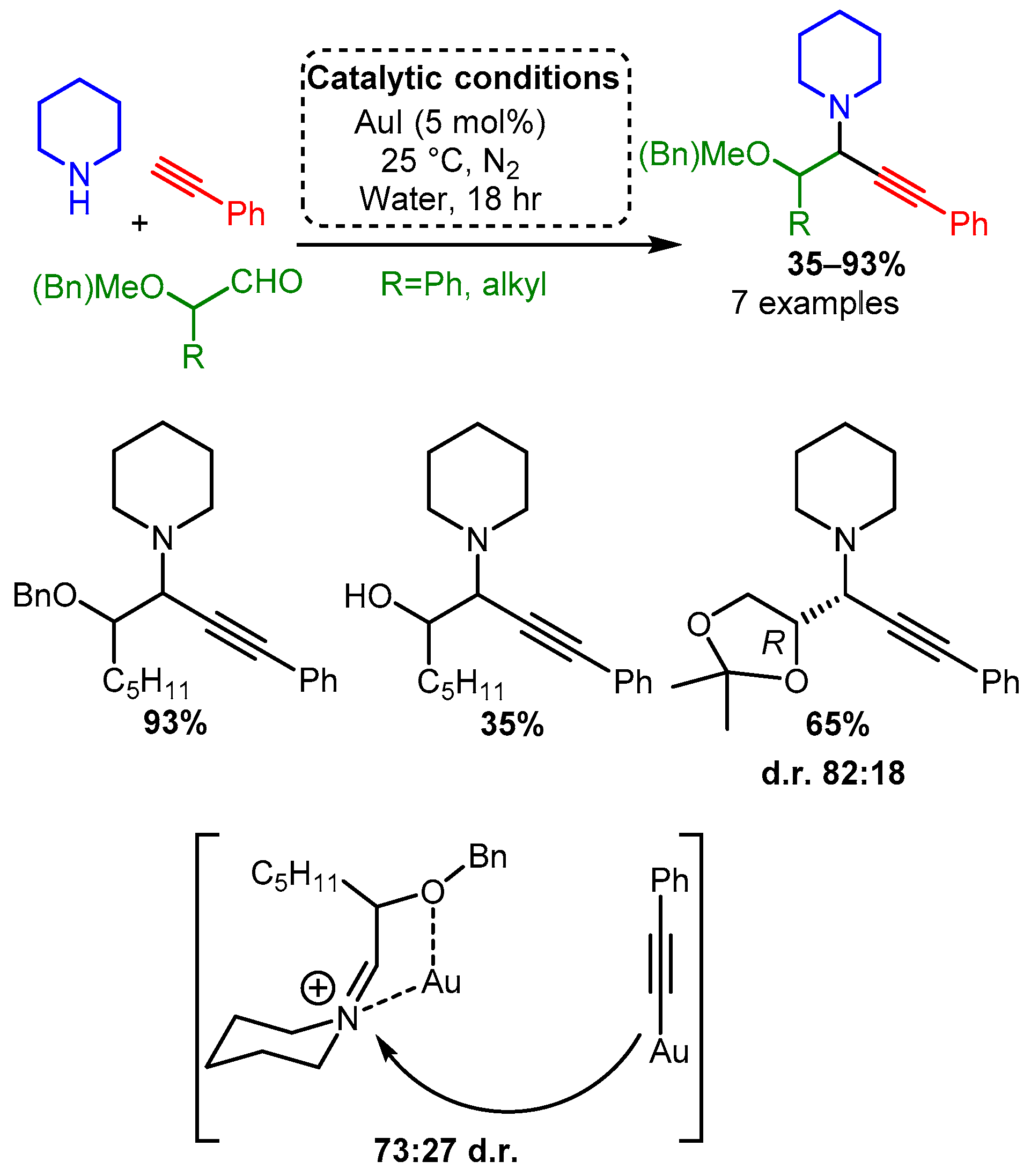 Scheme 17. Au(I) catalysed A3 reaction of α-oxyaldehydes and the proposed bimetallic mechanism that explains the observed diastereoselectivity.
Scheme 17. Au(I) catalysed A3 reaction of α-oxyaldehydes and the proposed bimetallic mechanism that explains the observed diastereoselectivity.Complimenting the reported homogenous catalysts, a heterogeneous gold catalyst was developed by Corma in which the partially charged and electron-deficient gold atoms were stabilised on nanocrystalline ZrO2 or CeO2 [70]. As mentioned before, this research focuses on homogeneous metal-based methodologies; therefore, Corma’s work is beyond the scope; however, it is interesting to note that these Au nanoparticles produced PAs in high yields, while also showing high diastereoselectivity when a chiral amine was used. In this research, a direct correlation was found between the formation of the Au(III) species, while no clear trend was observed for the Au(I) or Au(0) species. Notably, high yields were maintained even when an aliphatic alkyne or aldehyde were used together, although all amine substrates were secondary, and most were cyclic.
2.3. Other Metals
Of the metals outside group XI reported to catalyse the A3 coupling reaction, zinc and iron have been the most extensively studied, although indium, cadmium, cobalt, and nickel have all demonstrated promising catalytic activity. The use of a diethylzinc catalyst to alkynylated a C-N π bond was reported by Vallee, where the more electrophilic nitrones were used in place of imines to access N-propargyl-hydroxylamines in high yields [31]. The reaction required high catalyst loadings (20 mol%) and was carried out at room temperature under a nitrogen atmosphere. The reaction proceeded slowly in dichloromethane but was considerably improved using toluene, with time to completion varying from 1.5 to 30 h (Scheme 18).
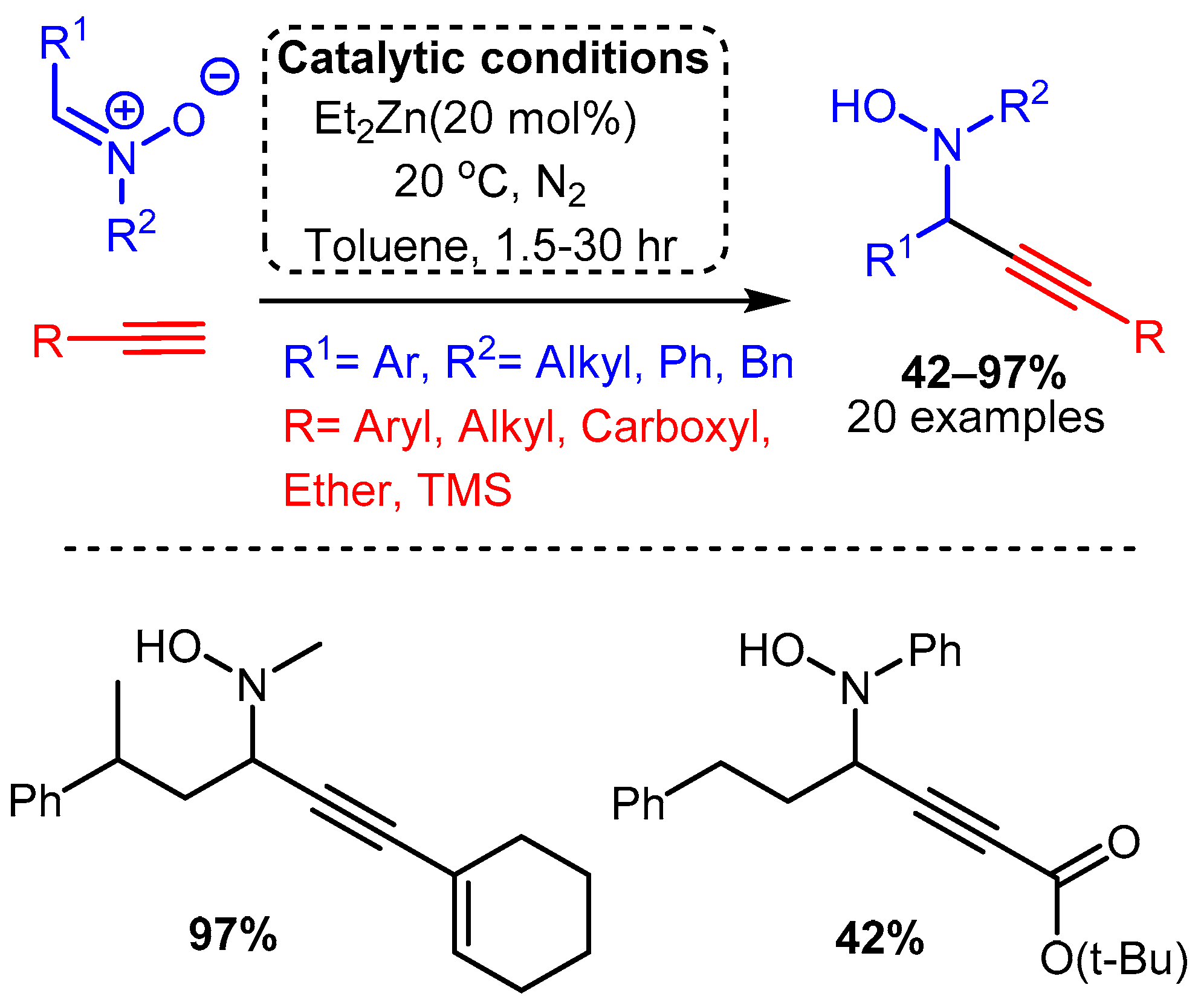 Scheme 18. Use of Et2Zn catalyst to add a terminal alkyne to a nitrone, as reported by Vallee and co-workers.
Scheme 18. Use of Et2Zn catalyst to add a terminal alkyne to a nitrone, as reported by Vallee and co-workers.A more traditional A3 coupling reaction was reported by Bolm, who first used a dimethylzinc catalyst to alkynylate pre-formed and protected imines and then applied the catalyst to the three-component reaction [32]. High temperatures, reaction times of 24 h, and extremely high catalyst loadings (150 mol%) were necessary to alkynylate the protected electron-withdrawing imines (R1CH=N-PG). The three-component reaction, which was conducted at room temperature, required 48–96 h and even higher catalyst loadings of 250–350 mol%. Moreover, the reported yields varied considerably, with an average of 67% yield for the protected imines and 64% for the one-pot reaction (Scheme 19).
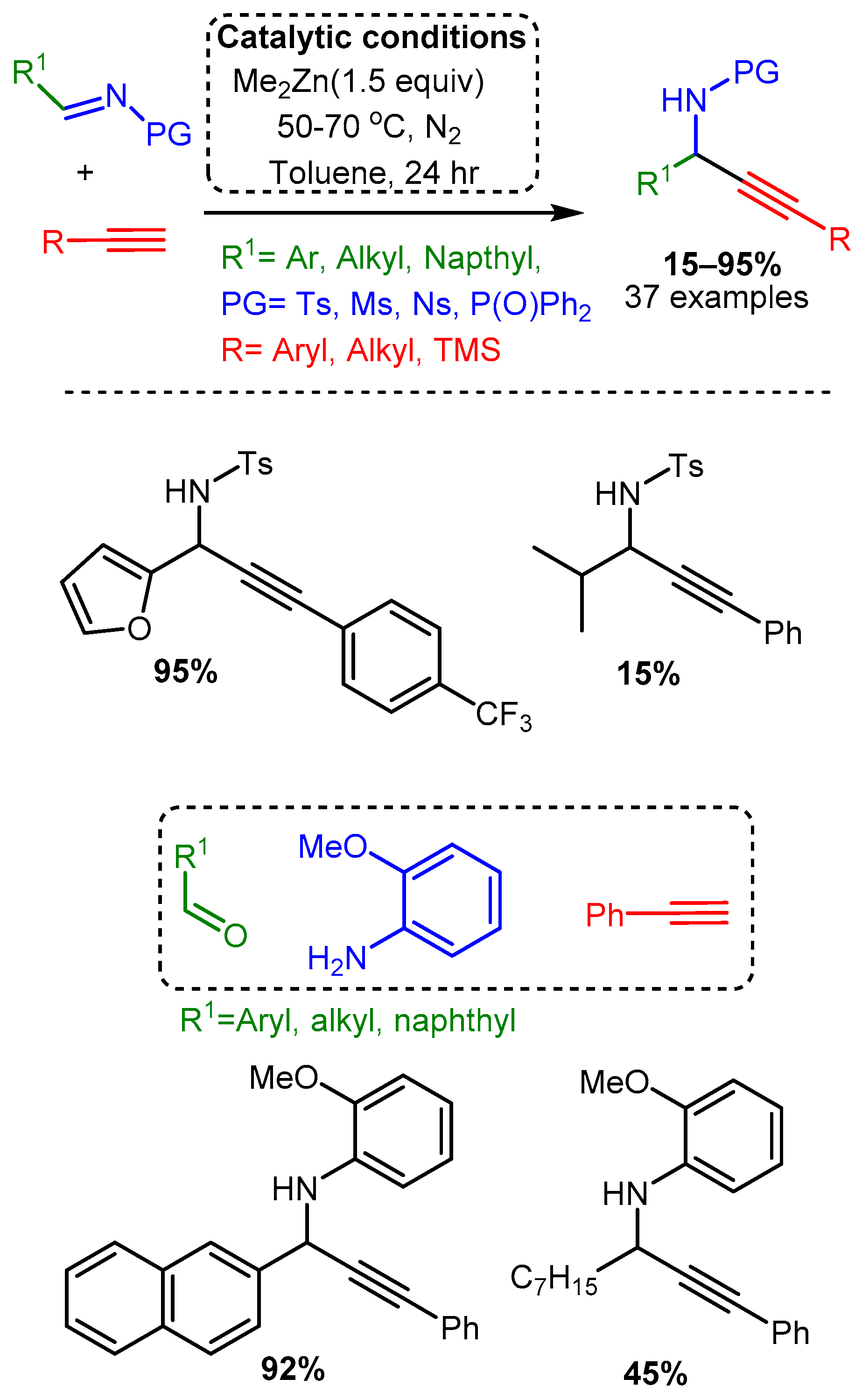 Scheme 19. Dimethylzinc-mediated alkynylation of imines for the synthesis protected propargylic amines.
Scheme 19. Dimethylzinc-mediated alkynylation of imines for the synthesis protected propargylic amines.More recent studies have focused on zinc salts in place of organic zinc derivatives. Chandak [34] explored the solvent-free A3 coupling reaction catalysed by Zn(OTf)2 in a study that lends itself to easy comparison to the above-discussed exploration of Cu(OTf)2 as a catalyst [68] (Scheme 14). The Zn(OTf)2 required lower catalytic loading (5 mol%) and catalysed the reaction in green, solvent-free conditions with high yields (Scheme 20). As with the Cu(OTf)2 catalyst, the electronic effects on the aromatic aldehyde did not alter the yield significantly. However, the zinc catalyst appears less sensitive to steric constraints than the copper catalyst. Of note, the zinc catalyst produced high yields when an aromatic alkyne was used, while the copper catalysts had the highest yields with an aliphatic alkyne; it should be mentioned, however, that the scope of alkyne used in both cases was limited.
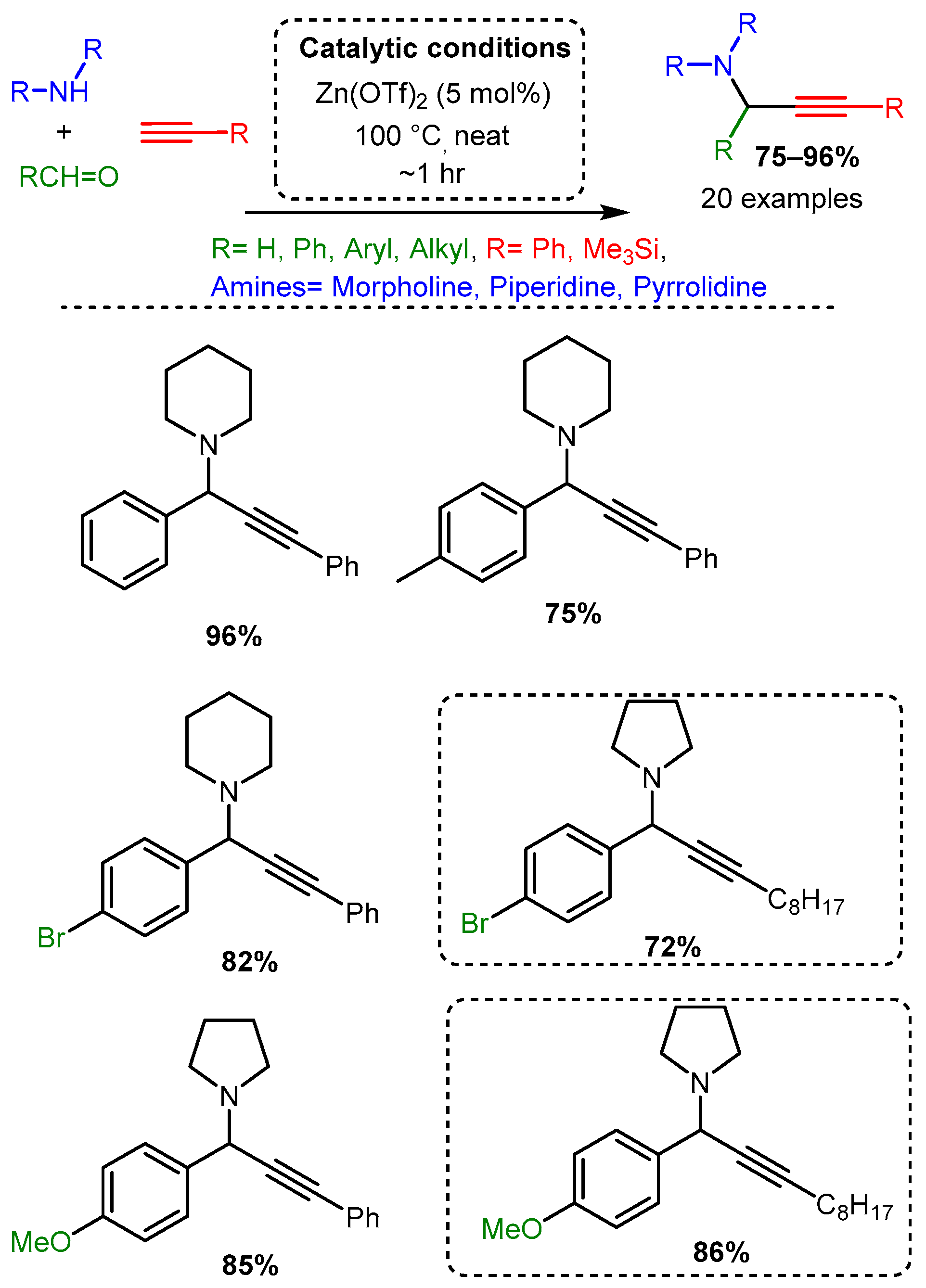 Scheme 20. Zn(OTf)2 catalysis of the A3 coupling reactions. Dashed line boxes denote comparable products from the Cu(OTf)2 catalysed reaction [65].
Scheme 20. Zn(OTf)2 catalysis of the A3 coupling reactions. Dashed line boxes denote comparable products from the Cu(OTf)2 catalysed reaction [65].Catalytic studies by Wang [71] and Li [25] have shown that PAs can be accessed in high yields by using FeCl3 in high loading (10 mol%). In both cases, the optimised reactions required high temperatures. However, the use of an inert atmosphere and toluene solvent by Wang and co-workers resulted in the corresponding PAs in average yields 10–20% higher than those achieved with solvent-free and open-air conditions in the later study of Li and co-workers (Scheme 21 and Scheme 22). These differences may be accounted for by variation in substrates, although it is challenging to conclude the exact substrate effects without further evidence.
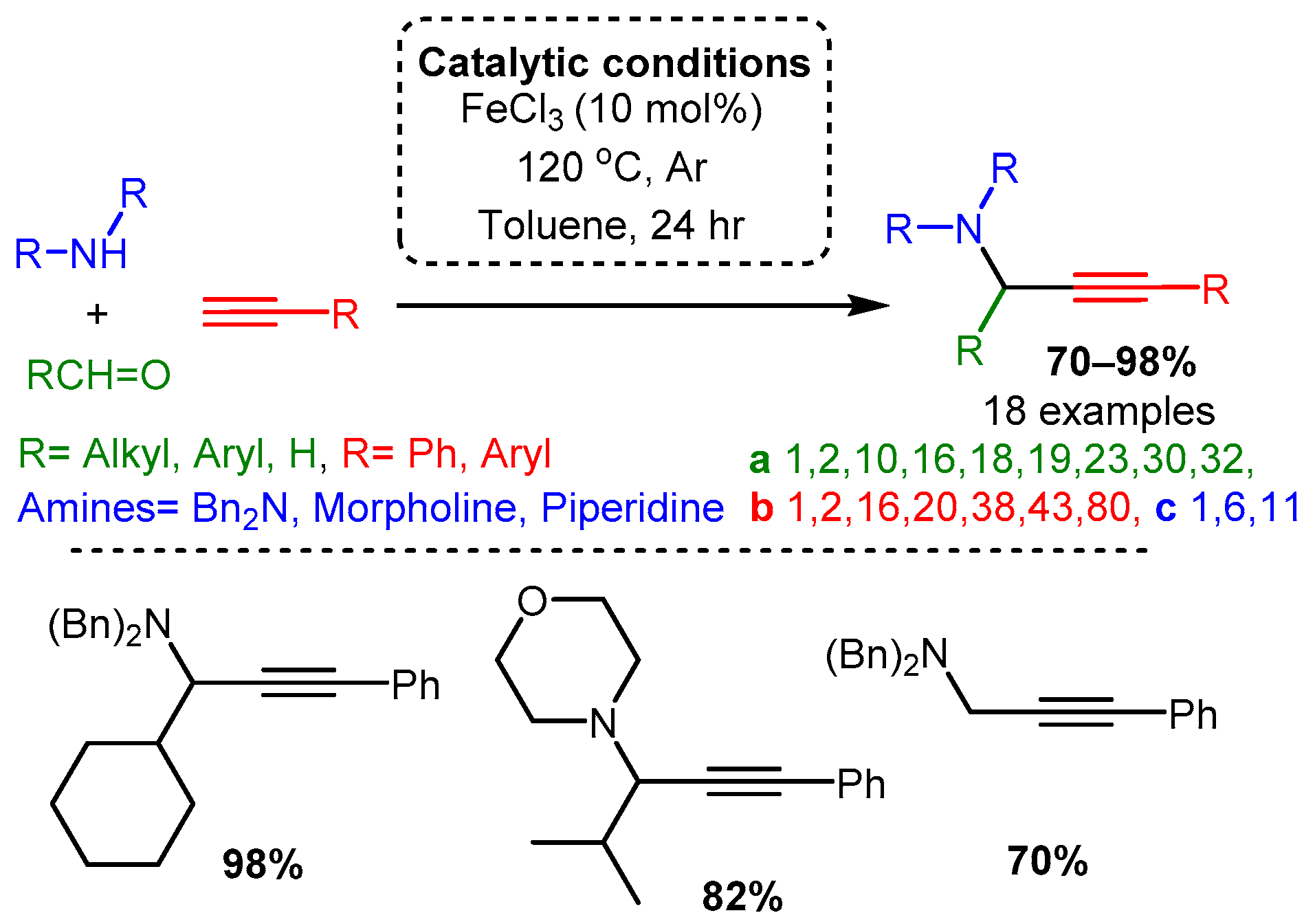 Scheme 21. Iron-catalysed A3 coupling reaction under argon atmosphere and toluene solvent.
Scheme 21. Iron-catalysed A3 coupling reaction under argon atmosphere and toluene solvent.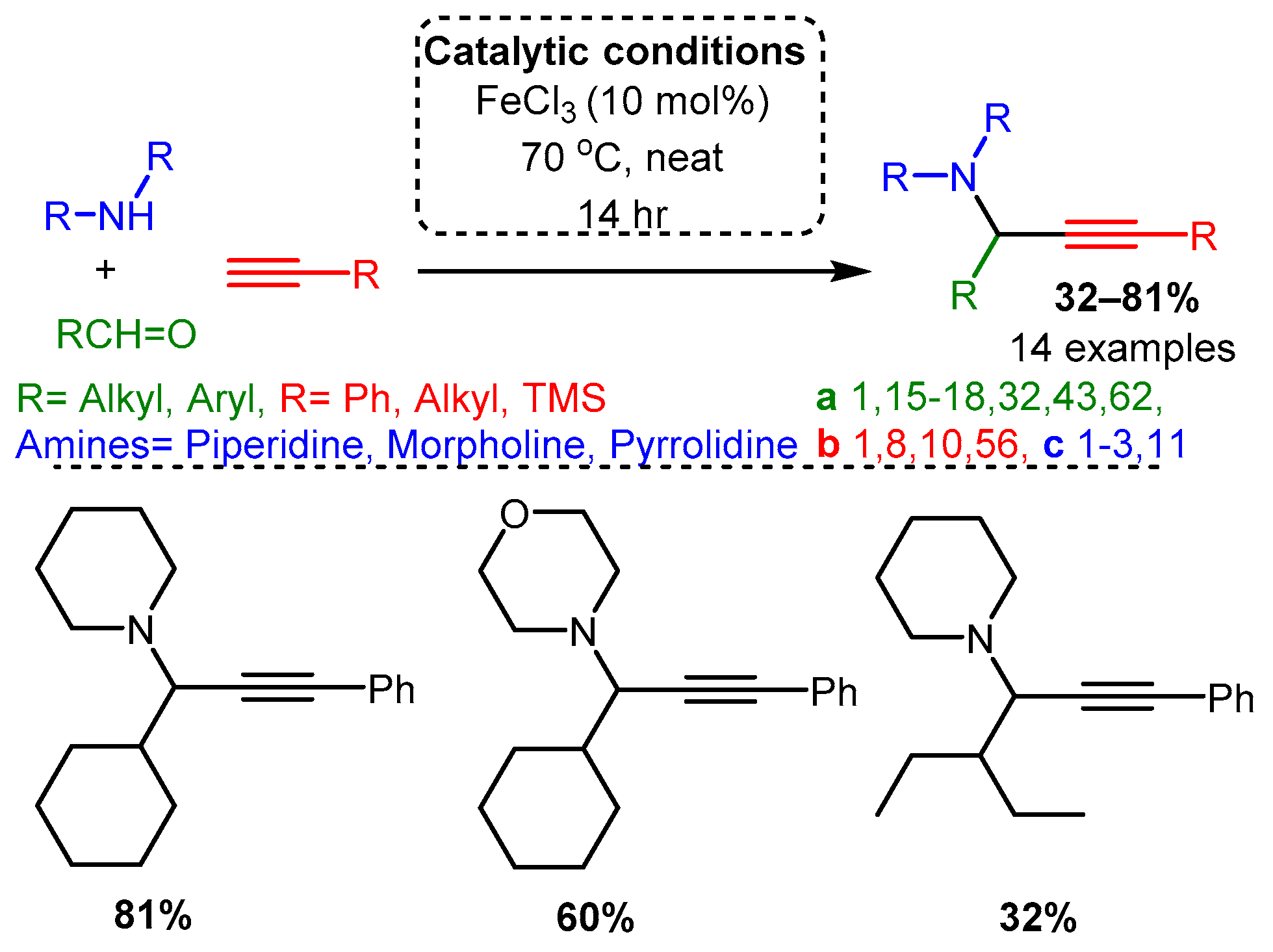 Scheme 22. Iron-catalysed A3 coupling reaction in open air and absence of solvent.
Scheme 22. Iron-catalysed A3 coupling reaction in open air and absence of solvent.2.4. Proposed In Situ Mechanisms
Significant advances have been made in understanding the catalytic mechanism using various organic ligands. Cu(I) and Cu(II) catalysts are most often employed in these studies due to their low cost, stability, and versatility. By understanding the catalytic mechanism, the hard–soft, HOMO–LUMO, and non-covalent interactions can be tailored to match specific combinations of catalyst, solvent, and substrates, thereby extending the applicability of the A3 coupling reaction and other metal-catalysed C-H activated additions. The proposed in situ catalytic cycles follow a similar path to the one in Scheme 23. This path involves the following steps:
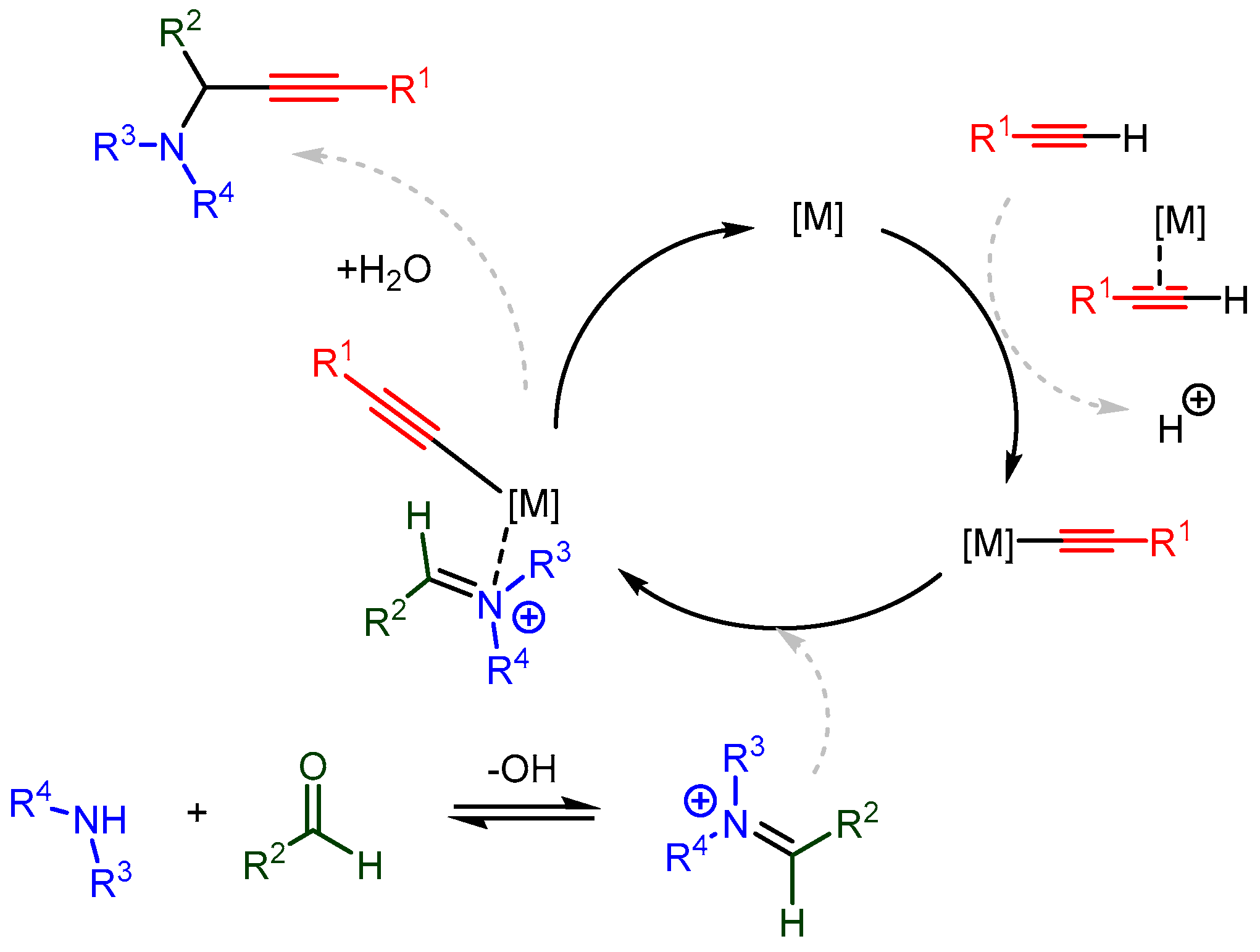 Scheme 23. The proposed in situ single metal catalysed mechanism for synthesizing PAs.
Scheme 23. The proposed in situ single metal catalysed mechanism for synthesizing PAs.-
A monomeric metal species forms a π-complex with the terminal alkyne;
-
The newly formed acetylide attacks the imine (or the iminium intermediate);
-
Decomplexation releases the PA and regenerates the metal catalyst.
References
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135.
- Bisai, V.; Suneja, A.; Singh, V.K. Asymmetric Alkynylation/Lactamization Cascade: An Expeditious Entry to Enantiomerically Enriched Isoindolinones. Angew. Chem. Int. Ed. 2014, 53, 10737–10741.
- Zhang, Y.; Huang, L.; Li, X.; Wang, L.; Feng, H. Chemo- and Diastereoselective Synthesis of N-Propargyl Oxazolidines through a Copper-Catalyzed Domino A3 Reaction. J. Org. Chem. 2019, 84, 5046–5055.
- Liu, C.; Wang, G.; Wang, Y.; Pereshivko, O.P.; Peshkov, V.A. Copper-Catalyzed Reaction of Secondary Propargylamines with Ethyl Buta-2,3-Dienoate for the Synthesis of 1,6-Dihydropyridines. European J. Org. Chem. 2019, 2019, 1981–1985.
- Singh, P.; Adolfsson, D.E.; Ådén, J.; Cairns, A.G.; Bartens, C.; Brännström, K.; Olofsson, A.; Almqvist, F. Pyridine-Fused 2-Pyridones via Povarov and A3 Reactions: Rapid Generation of Highly Functionalized Tricyclic Heterocycles Capable of Amyloid Fibril Binding. J. Org. Chem. 2019, 84, 3887–3903.
- Carmona, R.C.; Wendler, E.P.; Sakae, G.H.; Comassetoa, J.V.; Santos, A.A. Dos A3-Coupling Reaction as a Strategy towards the Synthesis of Alkaloids. J. Braz. Chem. Soc. 2015, 26, 117–123.
- Zhou, S.; Tong, R. Three-Step Catalytic Asymmetric Total Syntheses of 13-Methyltetrahydroprotoberberine Alkaloids. Org. Lett. 2017, 19, 1594–1597.
- Gommermann, N.; Knochel, P. Practical Highly Enantioselective Synthesis of Terminal Propargylamines. An Expeditious Synthesis of (S)-(+)-Coniine. Chem. Commun. 2004, 20, 2324–2325.
- Díez-González, S. Copper(I)–Acetylides: Access, Structure, and Relevance in Catalysis. In Advances in Organometallic Chemistry; Academic Press Inc.: Cambridge, MA, USA, 2016; Volume 66, pp. 93–141.
- Yamamoto, Y.; Gridnev, I.D.; Patil, N.T.; Jin, T. Alkyne Activation with Brønsted Acids, Iodine, or Gold Complexes, and Its Fate Leading to Synthetic Application. Chem. Commun. 2009, 5075–5087.
- Crabtree, R.H.; Lei, A. Introduction: CH Activation. Chem. Rev. 2017, 117, 8481–8482.
- Afewerki, S.; Córdova, A. Enamine/Transition Metal Combined Catalysis: Catalytic Transformations Involving Organometallic Electrophilic Intermediates. Top. Curr. Chem. 2019, 377, 38.
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569.
- Erkkilä, A.; Majander, I.; Pihko, P.M. Iminium Catalysis. Chem. Rev. 2007, 107, 5416–5470.
- Zou, Y.Q.; Hörmann, F.M.; Bach, T. Iminium and Enamine Catalysis in Enantioselective Photochemical Reactions. Chem. Soc. Rev. 2018, 47, 278–290.
- Innocenti, R.; Lenci, E.; Trabocchi, A. Recent Advances in Copper-Catalyzed Imine-Based Multicomponent Reactions. Tetrahedron Lett. 2020, 61, 152083.
- Jie, X.; Shang, Y.; Chen, Z.N.; Zhang, X.; Zhuang, W.; Su, W. Differentiation between Enamines and Tautomerizable Imines in the Oxidation Reaction with TEMPO. Nat. Commun. 2018, 9, 5002.
- Létinois-Halbes, U.; Pale, P.; Berger, S. Ag NMR as a Tool for Mechanistic Studies of Ag-Catalyzed Reactions: Evidence for in Situ Formation of Alkyn-1-Yl Silver from Alkynes and Silver Salts. J. Org. Chem. 2005, 70, 9185–9190.
- Fässler, R.; Tomooka, C.S.; Frantz, D.E.; Carreira, E.M. Infrared Spectroscopic Investigations on the Metallation of Terminal Alkynes by Zn(OTf)2. Proc. Natl. Acad. Sci. USA 2004, 101, 5843–5845.
- Wei, C.; Li, Z.; Li, C.J. The First Silver-Catalyzed Three-Component Coupling of Aldehyde, Alkyne, and Amine. Org. Lett. 2003, 5, 4473–4475.
- Wei, C.; Li, C.-J. A Highly Efficient Three-Component Coupling of Aldehyde, Alkyne, and Amines via C-H Activation Catalyzed by Gold in Water. J. Am. Chem. Soc. 2003, 125, 9584–9585.
- Li, C.J.; Wei, C. Highly Efficient Grignard-Type Imine Additions via C-H Activation in Water and under Solvent-Free Conditions. Chem. Commun. 2002, 2, 268–269.
- Raghuvanshi, D.S.; Singh, K.N. Highly Efficient Cadmium-Catalyzed Three-Component Coupling of an Aldehyde, Alkyne, and Amine via C-H Activation under Microwave Conditions. Synlett 2011, 3, 373–377.
- Chen, W.-W.; Bi, H.-P.; Li, C.-J. The First Cobalt-Catalyzed Transformation of Alkynyl C-H Bond: Aldehyde-Alkyne-Amine (A3) Coupling. Synlett 2010, 3, 475–479.
- Chen, W.W.; Nguyen, R.V.; Li, C.J. Iron-Catalyzed Three-Component Coupling of Aldehyde, Alkyne, and Amine under Neat Conditions in Air. Tetrahedron Lett. 2009, 50, 2895–2898.
- Li, P.H.; Wang, L. Mercurous Chloride Catalyzed Mannich Condensation of Terminal Alkynes with Secondary Amines and Aldehydes. Chin. J. Chem. 2005, 23, 1076–1080.
- Irfana Jesin, C.P.; Nandi, G.C. Catalyst-Controlled Dual Reactivity of Sulfonimidamides: Synthesis of Propargylamines and N-Propargyl Sulfonimidamides. Chem. Eur. J. 2019, 25, 743–749.
- Samai, S.; Nandi, G.C.; Singh, M.S. An Efficient and Facile One-Pot Synthesis of Propargylamines by Three-Component Coupling of Aldehydes, Amines, and Alkynes via C-H Activation Catalyzed by NiCl2. Tetrahedron Lett. 2010, 51, 5555–5558.
- Traverse, J.F.; Hoveyda, A.H.; Snapper, M.L. Enantioselective Synthesis of Propargylamines through Zr-Catalyzed Addition of Mixed Alkynylzinc Reagents to Arylimines. Org. Lett. 2003, 5, 3273–3275.
- Blay, G.; Cardona, L.; Climent, E.; Pedro, J.R. Highly Enantioselective Zinc/Binol-Catalyzed Alkynylation of N-Sulfonyl Aldimines. Angew. Chem. Int. Ed. 2008, 47, 5593–5596.
- Pinet, S.; Pandya, S.U.; Chavant, P.Y.; Ayling, A.; Vallee, Y. Dialkylzinc-Assisted Alkynylation of Nitrones. Org. Lett. 2002, 4, 1463–1466.
- Zani, L.; Alesi, S.; Cozzi, P.G.; Bolm, C. Dimethylzinc-Mediated Alkynylation of Imines. J. Org. Chem. 2006, 71, 1558–1562.
- Ying, J.; Wu, X.D.; Wang, D.; Pu, L. Catalytic Asymmetric Addition of Alkyl and Aryl Alkynes to N-(Diphenylphosphinoyl)Imines. J. Org. Chem. 2016, 81, 8900–8905.
- Sarode, P.B.; Bahekar, S.P.; Chandak, H.S. Zn(OTf)2-Mediated Expeditious and Solvent-Free Synthesis of Propargylamines via C-H Activation of Phenylacetylene. Synlett 2016, 27, 2209–2212.
- Wei, C.; Li, C.J. Enantioselective Direct-Addition of Terminal Alkynes to Imines Catalyzed by Copper(I)Pybox Complex in Water and in Toluene. J. Am. Chem. Soc. 2002, 124, 5638–5639.
- Bariwal, J.B.; Ermolat’Ev, D.S.; Van Der Eycken, E.V. Efficient Microwave-Assisted Synthesis of Secondary Alkylproparglamines by Using A3-Coupling with Primary Aliphatic Amines. Chem. Eur. J. 2010, 16, 3281–3284.
- Ohara, M.; Hara, Y.; Ohnuki, T.; Nakamura, S. Direct Enantioselective Three-Component Synthesis of Optically Active Propargylamines in Water. Chem. Eur. J. 2014, 20, 8848–8851.
- Ji, J.X.; Wu, J.; Chan, A.S.C. Catalytic Asymmetric Alkynylation of α-Imino Ester: A Versatile Approach to Optically Active Unnatural α-Amino Acid Derivatives. Proc. Natl. Acad. Sci. USA 2005, 102, 11196–11200.
- Dhanasekaran, S.; Kannaujiya, V.K.; Biswas, R.G.; Singh, V.K. Enantioselective A3-Coupling Reaction Employing Chiral CuI-i PrpyboxdiPh/ N-Boc-(l)-Proline Complex under Cooperative Catalysis: Application in the Synthesis of (Indol-2-Yl)Methanamines. J. Org. Chem. 2019, 84, 3275–3292.
- Koradin, C.; Polborn, K.; Knochel, P. Enantioselective Synthesis of Propargylamines by Copper-Catalyzed Addition of Alkynes to Enamines. Angew. Chem. Int. Ed. 2002, 41, 2535–2538.
- Sreenath, K.; Yuan, Z.; Macias-Contreras, M.; Ramachandran, V.; Clark, R.J.; Zhu, L. Dual Role of Acetate in Copper(II) Acetate Catalyzed Dehydrogenation of Chelating Aromatic Secondary Amines: A Kinetic Case Study of Copper-Catalyzed Oxidation Reactions. Eur. J. Inorg. Chem. 2016, 2016, 3728–3743.
- Park, K.; Heo, Y.; Lee, S. Metal-Free Decarboxylative Three-Component Coupling Reaction for the Synthesis of Propargylamines. Org. Lett. 2013, 15, 3322–3325.
- Loukopoulos, E.; Kallitsakis, M.; Tsoureas, N.; Abdul-Sada, A.; Chilton, N.F.; Lykakis, I.N.; Kostakis, G.E. Cu(II) Coordination Polymers as Vehicles in the A3 Coupling. Inorg. Chem. 2017, 56, 4898–4910.
- Yu, C.X.; Hu, F.L.; Liu, M.Y.; Zhang, C.W.; Lv, Y.H.; Mao, S.K.; Liu, L.L. Construction of Four Copper Coordination Polymers Derived from a Tetra-Pyridyl-Functionalized CalixArene: Synthesis, Structural Diversity, and Catalytic Applications in the A3 (Aldehyde, Alkyne, and Amine) Coupling Reaction. Cryst. Growth Des. 2017, 17, 5441–5448.
- Li, P.; Liu, Y.; Wang, L.; Xiao, J.; Tao, M. Copper(II)-Schiff Base Complex-Functionalized Polyacrylonitrile Fiber as a Green Efficient Heterogeneous Catalyst for One-Pot Multicomponent Syntheses of 1,2,3-Triazoles and Propargylamines. Adv. Synth. Catal. 2018, 360, 1673–1684.
- Naeimi, H.; Moradian, M. Copper(I)-N2S2-Salen Type Complex Covalently Anchored onto MCM-41 Silica: An Efficient and Reusable Catalyst for the A3-Coupling Reaction toward Propargylamines. Appl. Organomet. Chem. 2013, 27, 300–306.
- McDonagh, C.; O’Conghaile, P.; Klein Gebbink, R.J.M.; O’Leary, P. Electrostatic Immobilisation of Copper(I) and Copper(II) Bis(Oxazolinyl)Pyridine Catalysts on Silica: Application to the Synthesis of Propargylamines via Direct Addition of Terminal Alkynes to Imines. Tetrahedron Lett. 2007, 48, 4387–4390.
- Chen, G.J.; Chen, C.Q.; Li, X.T.; Ma, H.C.; Dong, Y. Bin Cu3L2 Metal-Organic Cages for A3-Coupling Reactions: Reversible Coordination Interaction Triggered Homogeneous Catalysis and Heterogeneous Recovery. Chem. Commun. 2018, 54, 11550–11553.
- Sun, W.J.; Gao, E.Q. MIL-101 Supported Highly Active Single-Site Metal Catalysts for Tricomponent Coupling. Appl. Catal. A Gen. 2019, 569, 110–116.
- Sun, W.J.; Xi, F.G.; Pan, W.L.; Gao, E.Q. MIL-101(Cr)-SO3Ag: An Efficient Catalyst for Solvent-Free A3 Coupling Reactions. J. Mol. Catal. A Chem. 2016, 430, 36–42.
- Li, Z.; Jiang, Z.; Su, W. Fast, Solvent-Free, Highly Enantioselective Three-Component Coupling of Aldehydes, Alkynes, and Amines Catalysed by the Copper(II)Pybox Complex under High-Vibration Ball-Milling. Green Chem. 2015, 17, 2330–2334.
- Sagadevan, A.; Pampana, V.K.K.; Hwang, K.C. Copper Photoredox Catalyzed A3’ Coupling of Arylamines, Terminal Alkynes, and Alcohols through a Hydrogen Atom Transfer Process. Angew. Chem. Int. Ed. 2019, 58, 3838–3842.
- Karthikeyan, P.; Arunrao, A.S.; Narayan, M.P.; Kumar, S.; Kumar, S.S.; Bhagat, P.R. Novel 1-Glycyl-3-Methyl Imidazolium Chloride-Iron(III) Complex for Synthesis of Propargylamines. Asian J. Chem. 2012, 24, 4285–4289.
- Esfandiary, N.; Pazoki, F.; Nakisa, A.; Azizi, K.; Radfar, I.; Heydari, A. Silver Chloride Supported on Vitamin B1 -Organometallic Magnetic Catalyst: Synthesis, Density Functional Theory Study and Application in A3-Coupling Reactions. Appl. Organomet. Chem. 2020, 34, e5725.
- Mirabedini, M.; Motamedi, E.; Kassaee, M.Z. Magnetic CuO Nanoparticles Supported on Graphene Oxide as an Efficient Catalyst for A3-Coupling Synthesis of Propargylamines. Chin. Chem. Lett. 2015, 26, 1085–1090.
- Paioti, P.H.S.; Abboud, K.A.; Aponick, A. Incorporation of Axial Chirality into Phosphino-Imidazoline Ligands for Enantioselective Catalysis. ACS Catal. 2017, 7, 2133–2138.
- Cardoso, F.S.P.; Abboud, K.A.; Aponick, A. Design, Preparation, and Implementation of an Imidazole-Based Chiral Biaryl P,N-Ligand for Asymmetric Catalysis. J. Am. Chem. Soc. 2013, 135, 14548–14551.
- Dube, H.; Gommermann, N.; Knochel, P. Synthesis of Chiral α-Aminoalkylpyrimidines Using an Enantioselective Three-Component Reaction. Synthesis 2004, 4, 2015–2025.
- Rokade, B.V.; Guiry, P.J. Diastereofacial π-Stacking as an Approach to Access an Axially Chiral P,N-Ligand for Asymmetric Catalysis. ACS Catal. 2017, 7, 2334–2338.
- Zhao, C.; Seidel, D. Enantioselective A3 Reactions of Secondary Amines with a Cu(I)/Acid-Thiourea Catalyst Combination. J. Am. Chem. Soc. 2015, 137, 4650–4653.
- Syeda Huma, H.Z.; Halder, R.; Singh Kalra, S.; Das, J.; Iqbal, J. Cu(I)-Catalyzed Three Component Coupling Protocol for the Synthesis of Quinoline Derivatives. Tetrahedron Lett. 2002, 43, 6485–6488.
- Gurubrahamam, R.; Periasamy, M. Copper(I) Halide Promoted Diastereoselective Synthesis of Chiral Propargylamines and Chiral Allenes Using 2-Dialkylaminomethylpyrrolidine, Aldehydes, and 1-Alkynes. J. Org. Chem. 2013, 78, 1463–1470.
- Zhang, Y.; Feng, H.; Liu, X.; Huang, L. A Highly Chemoselective Synthesis of Cyclic Divalent Propargylamines by Copper-Catalyzed Annulation/Double A3-Couplings. Eur. J. Org. Chem. 2018, 2018, 2039–2046.
- Feng, H.; Zhang, Y.; Zhang, Z.; Chen, F.; Huang, L. Copper-Catalyzed Annulation/A3-Coupling Cascade: Diastereodivergent Synthesis of Sterically Hindered Monocyclic Oxazolidines Bearing Multiple Stereocenters. Eur. J. Org. Chem. 2019, 1931–1939.
- Choi, Y.J.; Jang, H.Y. Copper-Catalyzed A3-Coupling: Synthesis of 3-Amino-1,4-Diynes. Eur. J. Org. Chem. 2016, 2016, 3047–3050.
- Lim, J.; Park, K.; Byeun, A.; Lee, S. Copper-Catalyzed Decarboxylative Coupling Reactions for the Synthesis of Propargyl Amines. Tetrahedron Lett. 2014, 55, 4875–4878.
- Meyet, C.E.; Pierce, C.J.; Larsen, C.H. A Single Cu(II) Catalyst for the Three-Component Coupling of Diverse Nitrogen Sources with Aldehydes and Alkynes. Org. Lett. 2012, 14, 964–967.
- Martinez-Amezaga, M.; Giordano, R.A.; Prada Gori, D.N.; Permingeat Squizatto, C.; Giolito, M.V.; Scharovsky, O.G.; Rozados, V.R.; Rico, M.J.; Mata, E.G.; Delpiccolo, C.M.L. Synthesis of Propargylamines: Via the A3 Multicomponent Reaction and Their Biological Evaluation as Potential Anticancer Agents. Org. Biomol. Chem. 2020, 18, 2475–2486.
- Huang, B.; Yao, X.; Li, C.J. Diastereoselective Synthesis of α-Oxyamines via Gold-, Silver- and Copper-Catalyzed, Three-Component Couplings of α-Oxyaldehydes, Alkynes, and Amines in Water. Adv. Synth. Catal. 2006, 348, 1528–1532.
- Zhang, X.; Corma, A. Supported Gold(III) Catalysts for Highly Efficient Three-Component Coupling Reactions. Angew. Chem. Int. Ed. 2008, 47, 4358–4361.
- Li, P.; Zhang, Y.; Wang, L. Iron-Catalyzed Ligand-Free Three-Component Coupling Reactions of Aldehydes, Terminal Alkynes, and Amines. Chem. Eur. J. 2009, 15, 2045–2049.
More
Information
Subjects:
Chemistry, Applied
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.9K
Entry Collection:
Organic Synthesis
Revisions:
3 times
(View History)
Update Date:
04 Jul 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No