 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mingliang Zhang | -- | 2295 | 2022-06-23 15:19:18 | | | |
| 2 | Jessie Wu | -2 word(s) | 2293 | 2022-06-30 03:38:03 | | | | |
| 3 | Jessie Wu | Meta information modification | 2293 | 2022-06-30 03:44:47 | | |
Video Upload Options
Most of the major retinal degenerative diseases are associated with significant levels of oxidative stress. One of the major sources contributing to the overall level of stress is the reactive oxygen species (ROS) generated by mitochondria. The driving force for ROS production is the proton gradient across the inner mitochondrial membrane. This gradient can be modulated by members of the uncoupling protein family, particularly the widely expressed UCP2. The overexpression and knockout studies of UCP2 in mice have established the ability of this protein to provide neuroprotection in a number of animal models of neurological disease, including retinal diseases. The expression and activity of UCP2 are controlled at the transcriptional, translational and post-translational levels, making it an ideal candidate for therapeutic intervention.
1. Transcriptional and Translational Regulation of the Uncoupling Protein 2 Gene
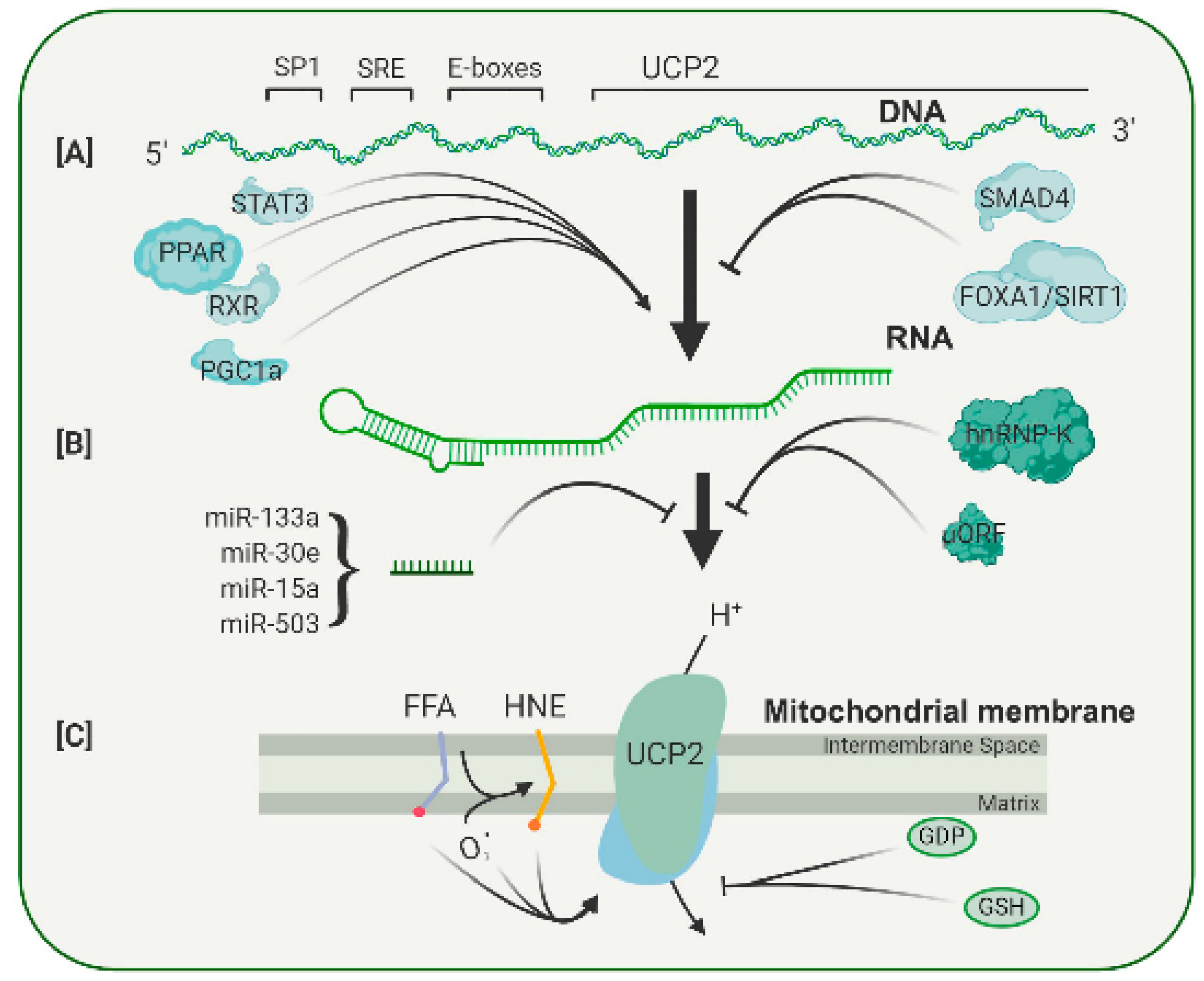
2. Pharmacological Regulation of Uncoupling Protein Activity
3. Uncoupling Proteins and Neural Degeneration
References
- Hass, D.T.; Barnstable, C.J. Uncoupling Proteins in the Mitochondrial Defense Against Oxidative Stress. Prog. Retin. Eye Res. 2021, 83, 100941.
- Lapp, D.W.; Zhang, S.S.; Barnstable, C.J. Stat3 Mediates LIF-induced Protection of Astrocytes Against Toxic ROS by Upregulating the UPC2 mRNA Pool. Glia 2014, 62, 159–170.
- He, Y.; Leung, K.W.; Ren, Y.; Pei, J.; Ge, J.; Tombran-Tink, J. PEDF Improves Mitochondrial Function in RPE Cells During Oxidative Stress. Investig. Opthalmol. Vis. Sci. 2014, 55, 6742–6755.
- Medvedev, A.V.; Snedden, S.K.; Raimbault, S.; Ricquier, D.; Collins, S. Transcriptional Regulation of the Mouse Uncoupling Protein-2 Gene. Double E-box Motif is Required for Peroxisome Proliferator-Activated Receptor-Gamma-Dependent Activation. J. Biol. Chem. 2001, 276, 10817–10823.
- Guan, J.; Zhao, M.; He, C.; Li, X.; Li, Y.; Sun, J.; Wang, W.; Cui, Y.L.; Zhang, Q.; Li, B.Y.; et al. Anti-Hypertensive Action of Fenofibrate via UCP2 Upregulation Mediated by PPAR Activation in Baroreflex Afferent Pathway. Neurosci. Bull. 2019, 35, 15–24.
- Chan, S.H.; Wu, K.L.; Kung, P.S.; Chan, J.Y. Oral Intake of Rosiglitazone Promotes a Central Antihypertensive Effect Via Upregulation of Peroxisome Proliferator-Activated Receptor-Gamma and Alleviation of Oxidative Stress in Rostral Ventrolateral Medulla of Spontaneously Hypertensive Rats. Hypertension 2010, 55, 1444–1453.
- Castrejon-Tellez, V.; Rodriguez-Perez, J.M.; Perez-Torres, I.; Perez-Hernandez, N.; Cruz-Lagunas, A.; Guarner-Lans, V.; Vargas-Alarcon, G.; Rubio-Ruiz, M.E. The Effect of Resveratrol and Quercetin Treatment On PPAR Mediated Uncoupling Protein (UCP-) 1, 2, and 3 Expression in Visceral White Adipose Tissue From Metabolic Syndrome Rats. Int. J. Mol. Sci. 2016, 17, 1069.
- Sayeed, A.; Meng, Z.; Luciani, G.; Chen, L.C.; Bennington, J.L.; Dairkee, S.H. Negative Regulation of UCP2 by TGFbeta Signaling Characterizes Low and Intermediate-Grade Primary Breast Cancer. Cell Death Dis. 2010, 1, e53.
- Graier, W.F.; Trenker, M.; Malli, R. Mitochondrial Ca2+, the Secret Behind the Function of Uncoupling Proteins 2 and 3? Cell Calcium 2008, 44, 36–50.
- Pecqueur, C.; Alves-Guerra, M.C.; Gelly, C.; Levi-Meyrueis, C.; Couplan, E.; Collins, S.; Ricquier, D.; Bouillaud, F.; Miroux, B. Uncoupling Protein 2, in Vivo Distribution, Induction upon Oxidative Stress, and Evidence for Translational Regulation. J. Biol. Chem. 2001, 276, 8705–8712.
- Brand, M.D.; Pakay, J.L.; Ocloo, A.; Kokoszka, J.; Wallace, D.C.; Brookes, P.S.; Cornwall, E.J. The Basal Proton Conductance of Mitochondria Depends On Adenine Nucleotide Translocase Content. Biochem. J. 2005, 392 Pt 2, 353–362.
- Ostrowski, J.; Klimek-Tomczak, K.; Wyrwicz, L.S.; Mikula, M.; Schullery, D.S.; Bomsztyk, K. Heterogeneous Nuclear Ribonucleoprotein K Enhances Insulin-Induced Expression of Mitochondrial UCP2 Protein. J. Biol. Chem. 2004, 279, 54599–54609.
- Hurtaud, C.; Gelly, C.; Bouillaud, F.; Levi-Meyrueis, C. Translation Control of UCP2 Synthesis by the Upstream Open Reading Frame. Cell. Mol. Life Sci. 2006, 63, 1780–1789.
- Rupprecht, A.; Moldzio, R.; Modl, B.; Pohl, E.E. Glutamine Regulates Mitochondrial Uncoupling Protein 2 to Promote Glutaminolysis in Neuroblastoma Cells. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 391–401.
- Hurtaud, C.; Gelly, C.; Chen, Z.; Levi-Meyrueis, C.; Bouillaud, F. Glutamine Stimulates Translation of Uncoupling Protein 2mRNA. Cell. Mol. Life Sci. 2007, 64, 1853–1860.
- Rubattu, S.; Stanzione, R.; Bianchi, F.; Cotugno, M.; Forte, M.; Della, R.F.; Fioriniello, S.; D’Esposito, M.; Marchitti, S.; Madonna, M.; et al. Reduced Brain UCP2 Expression Mediated by microRNA-503 Contributes to Increased Stroke Susceptibility in the High-Salt Fed Stroke-Prone Spontaneously Hypertensive Rat. Cell Death Dis. 2017, 8, e2891.
- Sun, L.L.; Jiang, B.G.; Li, W.T.; Zou, J.J.; Shi, Y.Q.; Liu, Z.M. MicroRNA-15a Positively Regulates Insulin Synthesis by Inhibiting Uncoupling Protein-2 Expression. Diabetes Res. Clin. Pract. 2011, 91, 94–100.
- Giardina, T.M.; Steer, J.H.; Lo, S.Z.; Joyce, D.A. Uncoupling Protein-2 Accumulates Rapidly in the Inner Mitochondrial Membrane During Mitochondrial Reactive Oxygen Stress in Macrophages. Biochim. Biophys. Acta 2008, 1777, 118–129.
- Rousset, S.; Mozo, J.; Dujardin, G.; Emre, Y.; Masscheleyn, S.; Ricquier, D.; Cassard-Doulcier, A.M. UCP2 is a Mitochondrial Transporter with an Unusual Very Short Half-Life. FEBS Lett. 2007, 581, 479–482.
- Azzu, V.; Affourtit, C.; Breen, E.P.; Parker, N.; Brand, M.D. Dynamic Regulation of Uncoupling Protein 2 Content in INS-1E Insulinoma Cells. Biochim. Biophys. Acta (BBA)—Bioenerg. 2008, 1777, 1378–1383.
- Schleiff, E.; McBride, H. The Central Matrix Loop Drives Import of Uncoupling Protein 1 Into Mitochondria. J. Cell Sci. 2000, 113 Pt 12, 2267–2272.
- Hansen, K.G.; Herrmann, J.M. Transport of Proteins Into Mitochondria. Protein J. 2019, 38, 330–342.
- Mailloux, R.J.; Seifert, E.L.; Bouillaud, F.; Aguer, C.; Collins, S.; Harper, M.E. Glutathionylation Acts as a Control Switch for Uncoupling Proteins UCP2 and UCP3. J. Biol. Chem. 2011, 286, 21865–21875.
- Mailloux, R.J.; Fu, A.; Robson-Doucette, C.; Allister, E.M.; Wheeler, M.B.; Screaton, R.; Harper, M.E. Glutathionylation State of Uncoupling Protein-2 and the Control of Glucose-Stimulated Insulin Secretion. J. Biol. Chem. 2012, 287, 39673–39685.
- Echtay, K.S.; Brand, M.D. Coenzyme Q Induces GDP-sensitive Proton Conductance in Kidney Mitochondria. Biochem. Soc. Trans. 2001, 29 Pt 6, 763–768.
- Berardi, M.J.; Shih, W.M.; Harrison, S.C.; Chou, J.J. Mitochondrial Uncoupling Protein 2 Structure Determined by NMR Molecular Fragment Searching. Nature 2011, 476, 109–113.
- Berardi, M.J.; Chou, J.J. Fatty Acid Flippase Activity of UCP2 is Essential for its Proton Transport in Mitochondria. Cell Metab. 2014, 20, 541–552.
- Pecqueur, C.; Bui, T.; Gelly, C.; Hauchard, J.; Barbot, C.; Bouillaud, F.; Ricquier, D.; Miroux, B.; Thompson, C.B. Uncoupling Protein-2 Controls Proliferation by Promoting Fatty Acid Oxidation and Limiting Glycolysis-Derived Pyruvate Utilization. FASEB J. 2008, 22, 9–18.
- Jaganjac, M.; Cindric, M.; Jakovcevic, A.; Zarkovic, K.; Zarkovic, N. Lipid Peroxidation in Brain Tumors. Neurochem. Int. 2021, 149, 105118.
- Gallo, G.; Sprovieri, P.; Martino, G. 4-Hydroxynonenal and Oxidative Stress in Several Organelles and its Damaging Effects on Cell Functions. J. Physiol. Pharmacol. 2020, 71, 15–33.
- Zhang, M.; Wang, L.; Wen, D.; Ren, C.; Chen, S.; Zhang, Z.; Hu, L.; Yu, Z.; Tombran-Tink, J.; Zhang, X.; et al. Neuroprotection of Retinal Cells by Caffeic Acid Phenylethyl Ester (CAPE) is Mediated by Mitochondrial Uncoupling Protein UCP2. Neurochem. Int. 2021, 151, 105214.
- Blanc, J.; Alves-Guerra, M.C.; Esposito, B.; Rousset, S.; Gourdy, P.; Ricquier, D.; Tedgui, A.; Miroux, B.; Mallat, Z. Protective Role of Uncoupling Protein 2 in Atherosclerosis. Circulation 2003, 107, 388–390.
- Deierborg, T.; Wieloch, T.; Diano, S.; Warden, C.H.; Horvath, T.L.; Mattiasson, G. Overexpression of UCP2 Protects Thalamic Neurons Following Global Ischemia in the Mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1186–1195.
- Mattiasson, G.; Shamloo, M.; Gido, G.; Mathi, K.; Tomasevic, G.; Yi, S.; Warden, C.H.; Castilho, R.F.; Melcher, T.; Gonzalez-Zulueta, M.; et al. Uncoupling Protein-2 Prevents Neuronal Death and Diminishes Brain Dysfunction After Stroke and Brain Trauma. Nat. Med. 2003, 9, 1062–1068.
- Diano, S.; Matthews, R.T.; Patrylo, P.; Yang, L.; Beal, M.F.; Barnstable, C.J.; Horvath, T.L. Uncoupling Protein 2 Prevents Neuronal Death Including that Occurring During Seizures: A Mechanism for Preconditioning. Endocrinology 2003, 144, 5014–5021.
- Sullivan, P.G.; Dube, C.; Dorenbos, K.; Steward, O.; Baram, T.Z. Mitochondrial Uncoupling Protein-2 Protects the Immature Brain From Excitotoxic Neuronal Death. Ann. Neurol. 2003, 53, 711–717.
- Andrews, Z.B. Uncoupling Protein-2 is Critical for Nigral Dopamine Cell Survival in a Mouse Model of Parkinson’s Disease. J. Neurosci. 2005, 25, 184–191.
- Hass, D.T.; Barnstable, C.J. Mitochondrial Uncoupling Protein 2 Knock-Out Promotes Mitophagy to Decrease Retinal Ganglion Cell Death in a Mouse Model of Glaucoma. J. Neurosci. 2019, 39, 3582–3596.
- Horvath, T.L.; Diano, S.; Leranth, C.; Garcia-Segura, L.M.; Cowley, M.A.; Shanabrough, M.; Elsworth, J.D.; Sotonyi, P.; Roth, R.H.; Dietrich, E.H.; et al. Coenzyme Q Induces Nigral Mitochondrial Uncoupling and Prevents Dopamine Cell Loss in a Primate Model of Parkinson’s Disease. Endocrinology 2003, 144, 2757–2760.
- Ramsden, D.B.; Ho, P.W.; Ho, J.W.; Liu, H.F.; So, D.H.; Tse, H.M.; Chan, K.H.; Ho, S.L. Human Neuronal Uncoupling Proteins 4 and 5 (UCP4 and UCP5): Structural Properties, Regulation, and Physiological Role in Protection Against Oxidative Stress and Mitochondrial Dysfunction. Brain Behav. 2012, 2, 468–478.
- Kwok, K.H.; Ho, P.W.; Chu, A.C.; Ho, J.W.; Liu, H.F.; Yiu, D.C.; Chan, K.H.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial UCP5 is Neuroprotective by Preserving Mitochondrial Membrane Potential, ATP Levels, and Reducing Oxidative Stress in MPP+ and Dopamine Toxicity. Free Radic. Biol. Med. 2010, 49, 1023–1035.
- Sanchis, D.; Fleury, C.; Chomiki, N.; Goubern, M.; Huang, Q.; Neverova, M.; Gregoire, F.; Easlick, J.; Raimbault, S.; Levi-Meyrueis, C.; et al. BMCP1, a Novel Mitochondrial Carrier with High Expression in the Central Nervous System of Humans and Rodents, and Respiration Uncoupling Activity in Recombinant Yeast. J. Biol. Chem. 1998, 273, 34611–34615.
- Mao, W.; Yu, X.X.; Zhong, A.; Li, W.; Brush, J.; Sherwood, S.W.; Adams, S.H.; Pan, G. UCP4, a Novel Brain-Specific Mitochondrial Protein that Reduces Membrane Potential in Mammalian Cells. FEBS Lett. 1999, 443, 326–330.
- Wei, Z.; Chigurupati, S.; Bagsiyao, P.; Henriquez, A.; Chan, S.L. The Brain Uncoupling Protein UCP4 Attenuates Mitochondrial Toxin-Induced Cell Death: Role of Extracellular Signal-Regulated Kinases in Bioenergetics Adaptation and Cell Survival. Neurotox. Res. 2009, 16, 14–29.
- Chu, A.C.; Ho, P.W.; Kwok, K.H.; Ho, J.W.; Chan, K.H.; Liu, H.F.; Kung, M.H.; Ramsden, D.B.; Ho, S.L. Mitochondrial UCP4 Attenuates MPP+—And Dopamine-Induced Oxidative Stress, Mitochondrial Depolarization, and ATP Deficiency in Neurons and is Interlinked with UCP2 Expression. Free Radic. Biol. Med. 2009, 46, 810–820.
- Mozo, J.; Ferry, G.; Studeny, A.; Pecqueur, C.; Rodriguez, M.; Boutin, J.A.; Bouillaud, F. Expression of UCP3 in CHO Cells Does Not Cause Uncoupling, but Controls Mitochondrial Activity in the Presence of Glucose. Biochem. J. 2006, 393 Pt 1, 431–439.

