1. Transcriptional and Translational Regulation of the Uncoupling Protein 2 Gene
The UCP2 gene extends over 8.4 kb and consists of eight exons and seven introns, a structure shared with the other members of the UCP family. While the intron/exon boundaries are conserved, there is considerable variation in intron size among the UCP genes, suggesting the evolution of different regulatory mechanisms. Following two 5′ untranslated exons, exons 3/4, 5/6 and 7/8 encode the three structural repeats of the protein. The structures of the UCP2 gene and protein have been reviewed in detail elsewhere
[1][2], but some features of this genomic structure are important to point out because they provide useful information about the regulation of UCP2 expression and activity.
Because UCP2 is expressed in most tissues, it contains promoter elements that respond to common transcription factors, such as SP1 (
Figure 1A). In addition, the UCP2 promoter contains a serum response element (SRE), which is likely to mediate the enhanced UCP2 transcription found after treatment with growth factors such as LIF and PEDF, both of which appear to act through a STAT3 signal pathway
[2][3][33,34]. In the UCP2 promoter, two E-boxes are also present, but the transcription factors binding to these have not yet been identified. The E-boxes do, however, appear to be involved in the binding of PPAR family of transcription factors
[4][35]. As shown in
Figure 1, there is evidence that PPAR agonists increase expression of UCP2 in many tissues
[5][6][36,37]. Treatment with the polyphenols resveratrol and quercetin caused an increase in levels of PPAR, and that, in turn, increased UCP2 RNA
[7][38]. In addition to the PPAR factors themselves, a PPAR cofactor, PGC-1, can also regulate UCP2 transcription. Because PGC-1 and the PPAR factors are involved in the regulation of genes active in a wide range of metabolic pathways, particularly in mitochondria, it seems that the levels of UCP2 transcription are coupled to the general metabolic state of a cell. In addition to these metabolic conditions, UCP2 transcription can be affected by growth factors, signifying another integrative level of control (
Figure 1). For example, TGF stimulates binding of SMAD4 to repressive elements in the UCP2 proximal promoter, and this decreases its transcription
[8][39]. On the other hand, PEDF increases the levels of UCP2 RNA and restores resistance to oxidative stress in aged RPE cells
[3][34]. The regulation of UCP2 transcription appears to be subtle, without dramatic increases or decreases, and there is growing evidence that the major control of UCP2 expression is through post-transcriptional mechanisms.
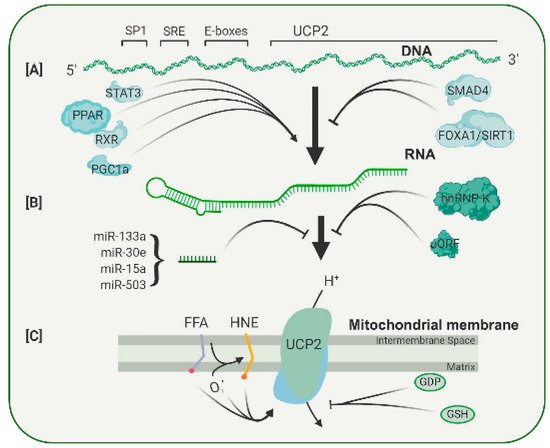
Figure 1. Diagrammatic representation of the multiple levels of regulation of UCP2 expression and activity. (
A) UCP2 transcription is enhanced or repressed through binding of several transcription factors to sites in its promoter. (
B) Translation of the UCP2 transcript is controlled by an upstream open reading frame (uORF), a number of microRNAs and RNS binding proteins, such as hnRNP-K. (
C) The UCP2 protein can be activated by a number of compounds, including free fatty acids and lipid peroxides, and can be inhibited by the nucleotide GDP and by glutathione. A more complete discussion of the regulation of UCP2 expression can be found in Reference
[1][2] from which this figure is adapted.
UCP2 is a relatively minor protein of mitochondria, and its expression varies considerably among tissues. The spleen has the highest level of UCP2, but even here, the amount has been estimated as 160-fold less than that of UCP1 in brown adipose tissue, constituting 0.01 to 0.1% of mitochondrial membrane protein in various tissues
[9][10][40,41]. Other tissues have up to 10-fold lower amounts of UCP2 than the spleen. This is in contrast with the ANT proteins that were found to be 6% of mitochondrial protein
[11][42]. The first indication of post-transcriptional control of UCP2 levels came from experiments in which the RNA and protein were measured in samples under different conditions. LPS treatment increased UCP2 protein levels by 12-fold without any measurable change in RNA levels
[10][41]. Since then, at least three post-transcriptional regulatory mechanisms have been described (
Figure 1B).
The first is a widely expressed RNA binding protein, hnRNP-K, which binds to sites in the 3′-UTR of UCP2 RNA. This binding is regulated by several factors, including insulin, angiopoietin and adiponectin, which can cross the blood–brain or blood–retina barrier and link UCP2 levels to the general metabolic status of the organism
[12][43].
The second regulatory mechanism operates at the 5′ untranslated region of the UCP2 RNA. Within this region, a 36-amino-acid open reading frame has been identified
[13][44]. This sequence has an inhibitory function, since mutations within the open reading frame lead to increased UCP2 protein expression. Activity of this uORF can be regulated by glutamine, a neurotransmitter precursor. Such regulation again suggests that UCP2 levels are regulated by the metabolic status of a cell
[14][15][45,46].
The third potential regulatory mechanism is less well defined. There is evidence that a number of microRNAs can alter UCP2 RNA levels in many tissues (
Figure 1B). Most studies show that stress or disease lowers the level of microRNA and leads to an increase in UCP2 protein
[16][47]. Many of these studies are correlative, although in a few cases, specific microRNAs were shown to bind to the 3′-untranslated UTR of UCP2 RNA
[17][48]. Some of these microRNAs are also found in the eye, but whether they play a role in UCP2 responses to retinal stress has yet to be determined.
One of the consequences of the complex post-transcriptional regulation of UCP2 is a rapid change in protein levels in response to modulations in the environment. This is primarily because the protein has a very short half-life. In several tissues, the half-life of UCP2 (and UCP3) has been measured as 1 h, considerably shorter than that of other inner mitochondrial membrane proteins, which have reported half-lives of between 4 and 17 days
[18][19][20][49,50,51]. Isolated mitochondria do not show rapid UCP2 turnover, so it is thought that protein degradation is regulated by specific cytoplasmic factors
[19][50]. Although these are not characterized, it is known that UCP2 enters the mitochondria via a series of specific chaperones and by a recognition sequence on UCP2 itself
[21][22][52,53]. The rapid turnover kinetics of UCP2 and the multiple post-transcriptional mechanisms that link its expression to cell metabolism both argue for a key role of this protein in mitochondrial function and its potential to combat disease and injury.
2. Pharmacological Regulation of Uncoupling Protein Activity
In general, there is a correlation between UCP2 expression and function (with overexpression leading to increased uncoupling activity). UCP2 is, however, regulated by a number of allosteric modulators
[1][2]. These include glutathione, mitochondrial matrix superoxide, GDP and fatty acids (
Figure 1C).
It was found that induction of glutathionylation with diamines in thymocytes inactivated proton leakage in a UCP2-specific manner. The primary site of glutathionylation is thought to be cys
256 based on studies with UCP3 and the conserved amino acid sequence
[23][54]. ROS were able to reverse this glutathionylation, indicating that ROS and glutathione act together to regulate mitochondrial proton leak and ROS levels. This process is also linked to glucose metabolism, as elevated glucose levels decreases glutathionylation and the UCP2-mediated proton leak
[24][55].
Regulation by GDP appears to be part of the network integrating ATP synthesis, mitochondrial membrane potential and ROS production
[25][56]. When nucleotide triphosphate concentrations are reduced, GDP concentrations are usually increased. When GDP inhibits UCP2, it increases the mitochondrial membrane potential and ATP synthesis. GDP binds inside the UCP2 cavity, and this allosterically dislocates a fatty acid molecule from its binding site, which in turn inhibits UCP2 activity
[26][20].
There is some debate about the role of fatty acids in regulating UCP2 activity. Many UCP2 models invoke a fatty acid molecule as the proton carrier that flips from side to side to give net proton flux
[27][57]. Some have suggested that fatty acid binding to UCP2 serves as an allosteric effector that participates in the shift in substrate preference between glucose and fatty acids as substrate choice
[28][29].
Of particular interest is the feedback regulation provided by the peroxidized fatty acid 4-hydroxynon-2-enal (4-HNE). 4-HNE is an aldehydic lipid peroxidation intermediate formed as a result of ROS interaction with unsaturated fatty acids and serves as a gauge of ROS production
[29][58]. 4-HNE can activate the UCP2-mediated proton leak and thus decrease ROS production. 4-HNE is toxic, and higher concentrations can often damage cells by forming adducts with many proteins through their reactive aldehyde groups and through covalent interactions with aminophospholipids
[30][59]. As described later, this prompted
our
esearchers successful search for 4-HNE mimetics to regulate UCP2 activity and provide protection for neurons from oxidative stresses. These mimetics are likely to be a source of therapeutics for a variety of neurodegenerative diseases.
3. Uncoupling Proteins and Neural Degeneration
Although acute and chronic neurological conditions may have different etiologies and symptoms and affect different brain regions, the end result of all of them is death of neurons. Oxidative stress has been identified as a major component of many of these conditions. In many such cases, degeneration occurs when the homeostatic mechanisms regulating ROS production and destruction are overwhelmed. Since a large component of the oxidative burden facing cells arises from ROS produced in mitochondria, this raises the interesting therapeutic possibility of enhancing these homeostatic mechanisms and providing cells with a greater tolerance of extrinsic oxidative stress. A strong case has been made for reducing mitochondrial stress and improving their function by using neuroprotective peptide factors, such as PEDF, and small molecules, such as CAPE
[3][31][34,60]. There is now a growing body of literature to support the idea that UCPs, particularly UCP2, can modulate mitochondrial ROS generation and that they can provide significant protection in a variety of diseases.
Apart from the thermogenic action of UCP1, the most intensively studied uncoupling protein is UCP2. There is a body of evidence showing that UCP2 is involved in many degenerative conditions, either directly through its uncoupling function or indirectly through its overall action on mitochondrial homeostasis. Some of the first evidence for a role for UCP2 came in studies of atherosclerosis where UCP2 knockout mice showed much higher oxidative stress and larger atherosclerotic plaques than wild-type mice
[32][61]. Conversely, mice overexpressing UCP2 were protected from the effects of global ischemia
[33][62]. Similarly, in UCP2 overexpressing mice, brain damage was reduced compared with wild-type mice in experimentally induced stroke
[34][63]. These same studies showed that UCP2 reduced cell death and caspase 3 activation induced by oxygen and glucose deprivation. Using a drug-induced model of grand mal seizures in mice,
reswe
archers were able to show that UCP2 overexpression reduced hippocampal neuron death
[35][64]. In younger wild-type animals, protection against seizure-induced excitotoxic cell death was achieved by using dietary methods (high fat) to increase the basal levels of UCP2
[36][65].
A well-established model of neurodegeneration involving oxidative stress is the degeneration of substantia nigra neurons induced by the neurotoxin MPTP. The active form of this compound, MPP+, rapidly produces symptoms that mirror many features of Parkinson’s disease. In a mouse model of Parkinson’s disease,
rwe
searchers found that transgenic mice overexpressing UCP2 retained significantly more nigral dopamine neurons and had higher striatal dopamine levels after MPTP treatment than wild type (36% cell loss vs. 62% loss in substantia nigra)
[37][66]. Conversely, UCP2 knockout mice showed approximately a two-fold reduction in dopamine neurons and dopamine levels than wild type following MPTP treatment. These data on cell loss correlated with the levels of ROS measured in nigral neurons, with overexpressing transgenics showing the lowest and UCP2 null mice the highest levels. I
n t
his study, it was also noted that UCP2 knockout mice had reduced mitochondria numbers. This was confirmed in a more recent study that indicated increased mitophagy in UCP2 knockout animals
[38][28]. These data argue that UCP2 activity plays an important role in maintaining mitochondrial numbers and quality. The studies on the mouse model of Parkinson’s disease confirmed
our
esearchers' earlier findings in a primate model where
researcherswe activated UCP2 by feeding the animals a UCP2 activator, Coenzyme Q
10 (CoQ
10)
[39][67]. This treatment induced uncoupling, as defined by increased state 4 respiration. Following administration of MPTP, the CoQ10-treated animals showed significantly lower loss of dopamine cells in the substantia nigra than untreated animals (13 vs. 74%). Though less direct than the mouse studies, the work with primates suggests that UCP2 activators are strong therapeutic candidates to treat Parkinson’s disease and possibly other neurological conditions.
While most of the current data for the involvement of uncoupling proteins in neurodegenerative disease come from studies on UCP2, there is some evidence that UCP4 and UCP5 may play a role in Parkinson’s disease as well. Both UCP4 and UCP5 are expressed in the brain, though with different levels in different brain regions
[40][68]. Overexpression of either UCP4 or UCP5 led to decreased mitochondrial membrane potential in neuronal cell lines and model organisms
[41][42][43][69,70,71]. UCP4 protected cultured SH-SY5Y cells from MMP+ toxicity and PC12 cells from 3-NP toxicity
[44][45][72,73]. Similarly, UCP5 protected against oxidative stress induced by MMP+ in SH-SY5Y cells
[41][69]. There is still debate about whether UCP4 and UCP5 act as true uncoupling proteins or proteins whose transport mechanisms are recruited to provide neuroprotection
[41][69]. Because UCP3 expression is confined to muscle, there are few studies associating it with disease. It appears that UCP3 is not involved in uncoupling but rather in the transport of a number of metabolic intermediates
[46][74].