Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Natalya S Zinkevich | -- | 2165 | 2022-06-28 16:41:39 | | | |
| 2 | Beatrix Zheng | Meta information modification | 2165 | 2022-06-29 03:19:10 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sylvester, A.L.; Zhang, D.X.; Ran, S.; Zinkevich, N.S. Role of NADPH Oxidases in Pathologies. Encyclopedia. Available online: https://encyclopedia.pub/entry/24589 (accessed on 24 July 2026).
Sylvester AL, Zhang DX, Ran S, Zinkevich NS. Role of NADPH Oxidases in Pathologies. Encyclopedia. Available at: https://encyclopedia.pub/entry/24589. Accessed July 24, 2026.
Sylvester, Anthony L., David X. Zhang, Sophia Ran, Natalya S. Zinkevich. "Role of NADPH Oxidases in Pathologies" Encyclopedia, https://encyclopedia.pub/entry/24589 (accessed July 24, 2026).
Sylvester, A.L., Zhang, D.X., Ran, S., & Zinkevich, N.S. (2022, June 28). Role of NADPH Oxidases in Pathologies. In Encyclopedia. https://encyclopedia.pub/entry/24589
Sylvester, Anthony L., et al. "Role of NADPH Oxidases in Pathologies." Encyclopedia. Web. 28 June, 2022.
Copy Citation
Nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases, NOX), were discovered in immune cells, such as neutrophils and macrophages, in the 1970s. Upon phagocytosis of pathogens, the enzymatic complex is activated and triggers O2− production in an “oxidative burst” that acts to kill pathogens. Over time, enzymes with a similar function located in various tissues have been identified and subsequently grouped into the NOX family of enzymes. The mitochondrial electron transport chain was soon demonstrated as another source of O2− due to a “leaky” electron transport system, its O2− scavenged by superoxide dismutase (SOD) into H2O2.
ROS
NADPH oxidase
NOX
1. Introduction
Release of reactive oxygen species (ROS), which are unstable oxygen-containing molecules that react easily with other molecules, is central to the maintenance of vascular homeostasis. The enzymatic sources of ROS include nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases, NOX), mitochondria, endothelial nitric oxide (NO) synthases, and xanthine oxidase (see Figure 1). ROS generation is regulated primarily by the NADPH oxidase (NOX) enzymes, which predominantly release hydrogen peroxide (H2O2) and superoxide (O2−). Since excessive superoxide production is implicated in numerous pathologies, such as vascular complications of diabetes [1], IPF [2] and PBC [3], NOX enzymes are of therapeutic interest as critical upstream inducers of H2O2 production.
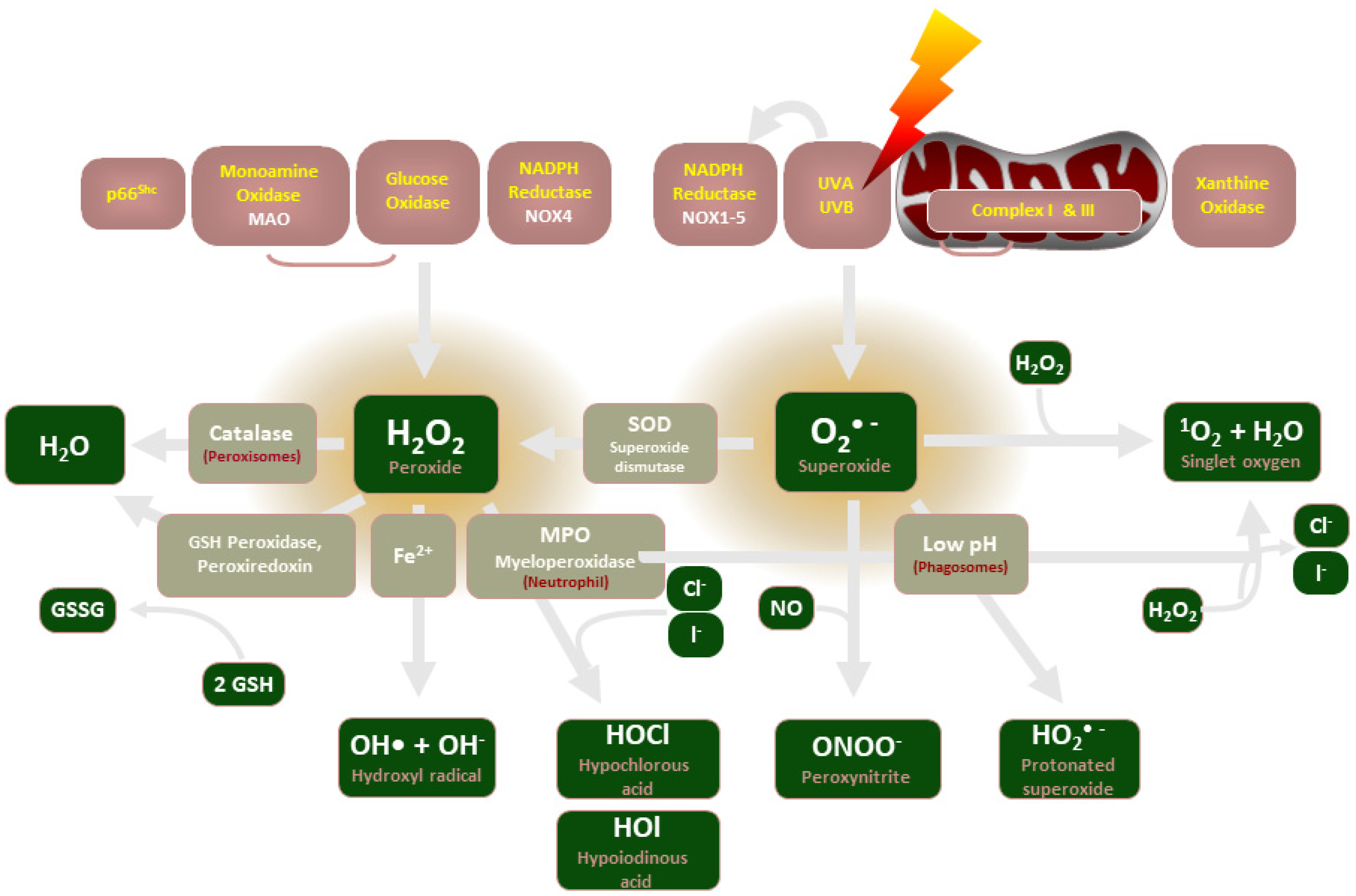
Figure 1. An illustration of the enzymes involved in ROS production and their common end-products.
2. NADPH Oxidase Description
Four NADPH oxidases (NOX1, NOX2, NOX4, and NOX5) are relevant to vascular homeostasis and pathology. NOX1 is widely distributed in various cell types, but expression is particularly abundant in the colonic epithelium and vascular smooth muscle cells [4]. It has been implicated in colon cancer progression [5] and vascular complications in diabetes [1]. The major ROS source in humans is NOX2 [6] that is highly expressed in phagocytes [7]. It is also the most widely expressed NOX isoform [8]. NOX2 contributes to endothelial dysfunction in vascular pathologies, such as insulin resistance in diabetes [9], but may also mediate phenotypic conversion of macrophages for tissue repair [10]. NOX4 is abundant in non-phagocytic cells, and it has been detected in vascular walls, fibroblasts, endothelial cells, and the kidney [8]. It mediates proinflammatory TGFβ-1 signaling in diseases such as IPF [8], but is also necessary for the polarization of macrophages [11]. NOX5 expression has been detected in lymphatic tissue, the testis, and blood vessels in humans [12]. Some studies showed that NOX5 contributes to vascular and kidney pathologies [12], but a recent study demonstrated a potential protective role of NOX5 against atherosclerosis in rabbits [13].
3. ROS and NADPH Oxidase in Recent Experimental Studies
Recent experimental data further highlight the role of NADPH oxidases and ROS in health and disease. Clinical intervention in illnesses of vascular dysfunction is limited by phenomena such as neointimal hyperplasia which drive further vascular damage. Palmitoylethanolamine (PEA) is a well- known anti-inflammatory agent and rutin (RUT) has antioxidant and vasoprotective properties. In a recent study, significant structural change in vessel morphology was observed following two weeks of carotid ligation, including ROS production and inflammatory cell infiltration [14]. Samples treated with a 1:1 ratio of PEA/RUT exhibited reduced change in vascular morphology, indicating that PEA/RUT administration was effective in attenuating inflammation, oxidative stress, vascular damage and vascular remodeling.
Thompson et al. demonstrated that microvascular function was restored in diet-induced obese mice following deletion of NOX1, independent of metabolic function [1], suggesting inhibition of NOX1 may be effective in preventing diabetic vascular complications. NOX1 is also highly expressed in colon cancer cells and supports their proliferation [5]. The 80–90% knockdown of NOX1 expression using shRNA increased tumor cell doubling time two- to three-fold without increasing apoptosis in HT-29 human colon cancer cells. A decline in hypoxia-inducible factor 1α (HIF-1α) downstream of attenuated NOX1 expression was associated with downregulation of mediators of cell proliferation and angiogenesis, including VEGF, c-MYC and c-MYB; the latter two are known oncogenes [15][16]. These data suggest that the proliferative phenotype in some colon cancers is supported by NOX1, and that NOX1 inhibition may be a therapeutic target for some colon cancers.
NADPH oxidases facilitate normal physiological processes by acting as signaling molecules. Recent studies have demonstrated their role in promoting macrophage polarization and subsequent tissue repair. The phenotypic conversion of pro-inflammatory Ly6ChiCX3CR1lo monocytes/macrophages to pro-resolving Ly6CloCX3CR1hi macrophages for liver repair is promoted by neutrophil signaling [10]. ROS, which are released by neutrophils via NOX2, are important mediators in this phenotypic conversion. Additionally, conversion was prevented in NOX2 deficient mice, indicating their role in tissue repair [7]. NOX4 has also recently been shown to be expressed in macrophages and to control their polarization in an NFκB-dependent manner [11]. NOX4 deficiency reduced the wound-healing M(IL4+IL13) macrophage population while forcing the polarization of proinflammatory M(LPS+IFNγ) macrophages. Decreased expression of NOX4 reduces STAT6 activation and promotes NFκB activity, resulting in increased NOX2 expression and associated O2− production. Thus, NOX4 also has an anti-inflammatory role in macrophages [7].
Previous work demonstrated that NOX4-derived H2O2 contributes to endothelium-dependent-vasodilation in mouse mesenteric arteries and rat intrarenal arteries [17][18] and that bradykinin (BK)-induced dilation in human arterioles depends on Nox-derived H2O2 [19]. A recent study demonstrated that NOX4 regulates the activity of TRPV4 endothelium-dependent dilation in human adipose and coronary arterioles via phosphorylation of Ser824 [20]. GKT137831, a NOX1/NOX4 inhibitor, reduced Ach-induced dilation as did the TRPV4 inhibitor HC067047, but GKT137831 did not further reduce vasodilation following application of HC067047, indicating a similar signaling pathway. These data support the authors’ novel hypothesis that NOX4-generated H2O2 stimulates phosphorylation, activating TRPV4 to cause Ca2+ influx and subsequent relaxation of the endothelium by factors such as NO.
NOX5 has been largely unstudied due to its absence in rodents. However, it is known to have functions in both homeostasis and pathogenesis. NOX5 contributes to coronary artery smooth muscle cell contraction, angiogenesis and atherosclerosis progression [21][22]. A recent study demonstrated a novel role for NOX5 in protecting against the progression of atherosclerosis [13]. NOX5-deficient young male rabbits consuming an atherogenic, high-fat feed developed significantly more plaques in the thoracic aorta compared to wild-type controls. These findings are in contradiction to previous work demonstrating that NOX5 drives atherosclerosis progression, highlighting its ill-studied nature. The mechanistic details of NOX5′s potentially protective role are unknown at this time and warrant further investigation.
4. NADPH Oxidases and ROS in Disease
NADPH oxidases have been implicated in numerous pathologies and pathology-driving pathways. Pulmonary epithelial cells express NOX isoforms including NOX1, NOX2, and NOX4 [23]. IPF is characterized by increased levels of mitochondrial and NADPH oxidase ROS. The condition is exacerbated by dysfunctional mitochondria which produce excess O2− and H2O2, increasing expression of NOX4 and TGFβ-1 signaling [2][24]. TGFβ-1 signaling promotes proinflammatory damage and collagen accumulation in the lungs [25], which in turn drives apoptosis and the formation of fibrotic tissue in IPF [26]. NOX4 additionally mediates the activity of TGFβ-1-induced cell differentiation, cardiac differentiation and transcriptional regulation [8]. Hepatocytes also express NOX isoforms, including NOX1, NOX2 and NOX4, which have been implicated in critical steps in initiating liver fibrosis, including hepatic stellate cell activation and hepatocyte apoptosis [3]. TGFβ-1 has also been demonstrated to contribute to the progression of PBC by enhancing fibrogenesis [27]. Together, these data suggest NADPH oxidases, particularly NOX4, may play a role in the progression of fibrotic pathologies, such as IPF and PBC, implicating selective NADPH oxidase inhibitors as promising therapeutic agents.
Gastroesophageal reflux disease (GERD) is a chronic digestive disease in which refluxate fluid frequently flows into the lower esophagus, causing esophagitis. GERD is the strongest known risk factor for esophageal adenocarcinoma [28]. Additionally, GERD has been implicated in the pathogenesis of IPF due to its high prevalence in IPF patients and the discovery of gastric acid components in bronchoalveolar lavage fluid of IPF patients [25][29]. Recent evidence suggests GERD pathogenesis is driven by an inflammatory environment constituted by increased production of cytokines, chemokines, ROS and a disturbed endogenous antioxidant defense system [25]. ROS generated by mitochondria and NOX1 and NOX2 have been demonstrated to contribute to the genotoxic effects of acidic bile reflux (BA/A). Elevated ROS production and associated DNA damage were attenuated with apocynin, NADPH oxidase-inhibiting gp91ds peptide, and siRNA targeting NOX1 and NOX2 [28]. These data suggest NOX1 and NOX2 inhibitors may be among promising treatments for esophageal adenocarcinomas.
The hallmarks of type 2 diabetes include hyperglycemia and insulin resistance. Insulin resistance is known to impair endothelium vasodilation and thus contributes to endothelial dysfunction [30]. ROS formation can be directly increased in tissues with hyperglycemia. Advanced glycation end-products that form in hyperglycemic tissues enhance the production of mitochondrial ROS and stimulate NADPH oxidases [9]. NADPH oxidases are further stimulated by the activation of protein kinase C and increased diacylglycerol synthesis in hyperglycemia [31]. NOX1 contributes to diabetic retinopathy [32], and NOX2 function has been demonstrated to be crucial in insulin resistance. The deletion of NOX2 in mice fed a high-fat diet significantly reduced insulin resistance [33], and O2− production in pulmonary endothelium in mutant insulin resistant mice was significantly reduced in NOX2-deficient subjects [30]. These data demonstrate the vital role of NOX2-produced ROS in insulin resistance, and further support the role of NADPH oxidases in driving endothelial dysfunction observed in type 2 diabetes.
Oxidative stress and endothelial dysfunction are additionally driven by angiotensin II (Ang II) signaling. Ang II contributes to atherosclerosis and, in hypertension, a major risk factor for cardiovascular and cerebrovascular disease, mediates functional and structural changes in vasculature [34][35]. NADPH oxidases are activated by Ang II, playing a role in the increase in ROS in vascular cells induced by Ang II. NOX2, in particular, may be a prominent mediator of Ang II-induced oxidative stress and its harmful effects in cerebral circulation during hypertension [8].
Regulated cell death plays an important role in the pathogenesis of conditions such as neurodegenerative diseases and cancer. Various regulated cell death pathways (ferroptosis, pyroptosis, necroptosis, alkilaptosis) may share common signals, such as redox signaling. Ferroptosis is a regulated necrosis resulting in the damage and rupture of the cell membrane driven by the accumulation of iron and subsequent lipid peroxidation by lipoxygenase (ALOX) or cytochrome P450 (POR). ALOX- and POR-mediated lipid peroxidation is promoted by NADPH oxidase, particularly NOX1, NOX2, and NOX4 and mitochondrial ROS [36]. Thus, NADPH oxidase inhibition for the purpose of attenuating regulated cell death pathways involved in cancer and neurodegenerative pathogenesis warrants further investigation.
5. NADPH Oxidase-Produced H2O2 Mediates Vascular Tone in Healthy Subjects during Exercise
A novel transition from an NO- to ROS-mediated mechanism of vasodilation has been described in human microcirculation. Nitric oxide synthases (NOS) produce NO, the regulator of vascular tone in healthy adults. During exercise, a switch to NADPH oxidase-produced H2O2 to mediate vasodilation occurs [37]. This is transient in healthy subjects but permanent in patients with coronary artery disease (CAD), a condition in which coronary arteries send insufficient blood to the heart, typically due to atherosclerosis and inflammation. The switch to H2O2-mediated vasodilation, in turn, perpetuates the chronic inflammatory condition in CAD. The mechanisms responsible for the transition in endothelial factors are not fully understood. NOS produces NO in healthy adults through the catalysis of L-arginine, O2 and NADPH-derived electrons. Tetrahydrobiopterin (BH4) is necessary for electron transfer during this process, and, if limited, NOS will instead catalyze the formation of O2−. Superoxide reduces the availability of BH4, thus perpetuating the loop of O2− production [9]. The formation of ONOO− from O2− reacting with NO further reduces the presence of NO and can both directly and indirectly drive oxidative stress [38]. This self-perpetuating cycle may, in turn, create a reliance on NADPH oxidase-produced H2O2 to preserve vascular tone [39].
6. RIRR, Endothelial Dysfunction and Angiogenesis
Endothelial dysfunction can also originate from several self-perpetuating ROS-producing loops. The formation of H2O2 and O2− is primarily modulated by NADPH oxidases, the mitochondrial electron transport chain, and xanthine oxidase [9]. NOX4-produced H2O2 stimulates NOX2 to produce O2−, which in turn stimulates mitochondria to produce O2− and H2O2 via the electron transport chain, a process termed ROS-induced ROS release (RIRR) [40]. ROS produced by mitochondria then activate NADPH oxidases [40] perpetuating this loop. ROS are also generated when hypoxanthine is oxidized to xanthine by xanthine oxidase, which is then converted to uric acid, forming H2O2 and O2− by transferring electrons to O2 [9]. These mechanisms ultimately perpetuate the inflammatory environment.
ROS-induced ROS release orchestrated by NOX4, NOX2, and mitochondria converts endothelial cells (EC) from a quiescent to an angiogenic phenotype by enhancing ROS-dependent VEGF signaling [41]. Either NOX4 or NOX2 knockdown or overexpression of mito-catalase (a scavenger of mitochondria-derived H2O2) inhibited EC migration and proliferation, thus providing evidence of a feed-forward mechanism responsible for driving the angiogenic process [41]. Elucidation of this signaling pathway would enable the development of promising therapeutic strategies aiming to modulate angiogenesis to alleviate various pathologies.
Peripheral arterial disease (PAD) is a circulatory condition in which occlusion of arteries supplying lower extremities depletes blood supply and may lead to amputation. Enhancing ROS-dependent VEGF-signaling may be used as an important therapeutic approach to promote neovascularization and tissue repair in PAD patients. On the other hand, inhibiting this signaling pathway may prevent excessive angiogenesis contributing to cancer, diabetic retinopathy, and atherosclerosis [42].
References
- Thompson, J.A.; Larion, S.; Mintz, J.D.; de Chantemèle, E.J.B.; Fulton, D.J.; Stepp, D.W. Genetic deletion of NADPH oxidase 1 rescues microvascular function in mice with metabolic disease. Circ. Res. 2017, 121, 502–511.
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115.
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17.
- Szanto, I.; Rubbia-Brandt, L.; Kiss, P.; Steger, K.; Banfi, B.; Kovari, E.; Herrmann, F.; Hadengue, A.; Krause, K.-H. Expression ofNOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathol. 2005, 207, 164–176.
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887.
- Lam, G.Y.; Huang, J.; Brumell, J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin. Immunopathol. 2010, 32, 415–430.
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991.
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814.
- Brown, O.I.; Bridge, K.I.; Kearney, M.T. Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Glucose Homeostasis and Diabetes-Related Endothelial Cell Dysfunction. Cells 2021, 10, 2315.
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076.
- Helfinger, V.; Palfi, K.; Weigert, A.; Schröder, K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFκB-Dependent Manner. Oxidative Med. Cell. Longev. 2019, 2019, 3264858.
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.D.L. NOX5: Molecular biology and pathophysiology. Exp. Physiol. 2019, 104, 605–616.
- Petheő, G.L.; Kerekes, A.; Mihálffy, M.; Donkó, Á.; Bodrogi, L.; Skoda, G.; Baráth, M.; Hoffmann, O.I.; Szeles, Z.; Balázs, B.; et al. Disruption of the NOX5 Gene Aggravates Atherosclerosis in Rabbits. Circ. Res. 2021, 128, 1320–1322.
- Fusco, R.; Siracusa, R.; Gugliandolo, E.; Peritore, A.F.; D’Amico, R.; Cordaro, M.; Crupi, R.; Impellizzeri, D.; Gomiero, C.; Cuzzocrea, S.; et al. Micro Composite Palmitoylethanolamide/Rutin Reduces Vascular Injury through Modulation of the Nrf2/HO−1 and NF-kB Pathways. Curr. Med. Chem. 2021, 28, 6287–6302.
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553.
- Fry, E.A.; Inoue, K. c-MYB and DMTF1 in Cancer. Cancer Investig. 2019, 37, 46–65.
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arter. Thromb. Vasc. Biol. 2011, 31, 1368–1376.
- Muñoz, M.; Martínez, M.P.; López-Oliva, M.E.; Rodríguez, C.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernández, M.; Rivera, L.; Medina, J.S.; et al. Hydrogen peroxide derived from NADPH oxidase 4- and 2 contributes to the endothelium-dependent vasodilatation of intrarenal arteries. Redox Biol. 2018, 19, 92–104.
- Larsen, B.T.; Bubolz, A.H.; Mendoza, S.A.; Pritchard, K.A.; Gutterman, D.D. Bradykinin-Induced Dilation of Human Coronary Arterioles Requires NADPH Oxidase–Derived Reactive Oxygen Species. Arter. Thromb. Vasc. Biol. 2009, 29, 739–745.
- Xie, Y.; Nishijima, Y.; Zinkevich, N.S.; Korishettar, A.; Fang, J.; Mathison, A.J.; Zimmermann, M.T.; Wilcox, D.A.; Gutterman, D.D.; Shen, Y.; et al. NADPH oxidase 4 contributes to TRPV4-mediated endothelium-dependent vasodilation in human arterioles by regulating protein phosphorylation of TRPV4 channels. Basic Res. Cardiol. 2022, 117, 24.
- Wang, H.; Hartnett, M.E. Roles of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase in Angiogenesis: Isoform-Specific Effects. Antioxidants 2017, 6, 40.
- Damico, R.; Zulueta, J.J.; Hassoun, P.M. Pulmonary Endothelial Cell NOX. Am. J. Respir. Cell Mol. Biol. 2012, 47, 129–139.
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827.
- Nelkine, L.; Vrolijk, M.F.; Drent, M.; Bast, A. Role of antioxidants in the treatment of gastroesophageal reflux disease-associated idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 2020, 26, 363–371.
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2012, 1832, 1028–1040.
- Liu, B.; Zhang, X.; Zhang, F.-C.; Zong, J.-B.; Zhang, W.; Zhao, Y. Aberrant TGF-β1 signaling contributes to the development of primary biliary cirrhosis in murine model. World J. Gastroenterol. 2013, 19, 5828–5836.
- Bhardwaj, V.; Gokulan, R.C.; Horvat, A.; Yermalitskaya, L.; Korolkova, O.; Washington, K.M.; El-Rifai, W.; Dikalov, S.I.; Zaika, A.I. Activation of NADPH oxidases leads to DNA damage in esophageal cells. Sci. Rep. 2017, 7, 9956.
- Gole, H.; Tharp, D.L.; Bowles, D.K. Upregulation of Intermediate-Conductance Ca2+-Activated K+ Channels (KCNN4) in Porcine Coronary Smooth Muscle Requires NADPH Oxidase 5 (NOX5). PLoS ONE 2014, 2, e105337.
- Minnis, P.; Henry, K.; Keane, M.P. Reflux in idiopathic pulmonary fibrosis: Table 1. QJM Int. J. Med. 2015, 109, 7–10.
- Sukumar, P.; Viswambharan, H.; Imrie, H.; Cubbon, R.M.; Yuldasheva, N.; Gage, M.; Galloway, S.; Skromna, A.; Kandavelu, P.; Santos, C.X.; et al. Nox2 NADPH Oxidase Has a Critical Role in Insulin Resistance–Related Endothelial Cell Dysfunction. Diabetes 2013, 62, 2130–2134.
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119.
- Wilkinson-Berka, J.L.; Deliyanti, D.; Rana, I.; Miller, A.G.; Agrotis, A.; Armani, R.; Szyndralewiez, C.; Wingler, K.; Touyz, R.; Cooper, M.E.; et al. NADPH Oxidase, NOX1, Mediates Vascular Injury in Ischemic Retinopathy. Antioxid. Redox Signal. 2014, 20, 2726–2740.
- De Figueiredo, A.S.P.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J. Biol. Chem. 2015, 290, 13427–13439.
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913.
- Chrissobolis, S.; Banfi, B.; Sobey, C.G.; Faraci, F.M. Role of nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J. Appl. Physiol. 2012, 113, 184–191.
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 969.
- Durand, M.J.; Dharmashankar, K.; Bian, J.T.; Das, E.; Vidovich, M.; Gutterman, D.D.; Phillips, S.A. Acute exertion elicits a H2O2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension 2015, 65, 140.
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848.
- Zinkevich, N.S.; Fancher, I.S.; Gutterman, D.D.; Phillips, S.A. Roles of NADPH oxidase and mitochondria in flow-induced vasodilation of human adipose arterioles: ROS-induced ROS release in coronary artery disease. Microcirculation 2017, 24, e12380.
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Circ. Physiol. 2011, 301, H647–H653.
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Physiol. 2017, 312, C749–C764.
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849.
More
Information
Subjects:
Cardiac & Cardiovascular Systems
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
29 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No