Nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases, NOX), were discovered in immune cells, such as neutrophils and macrophages, in the 1970s. Upon phagocytosis of pathogens, the enzymatic complex is activated and triggers O2− production in an “oxidative burst” that acts to kill pathogens. Over time, enzymes with a similar function located in various tissues have been identified and subsequently grouped into the NOX family of enzymes. The mitochondrial electron transport chain was soon demonstrated as another source of O2− due to a “leaky” electron transport system, its O2− scavenged by superoxide dismutase (SOD) into H2O2.
- ROS
- NADPH oxidase
- NOX
1. Introduction
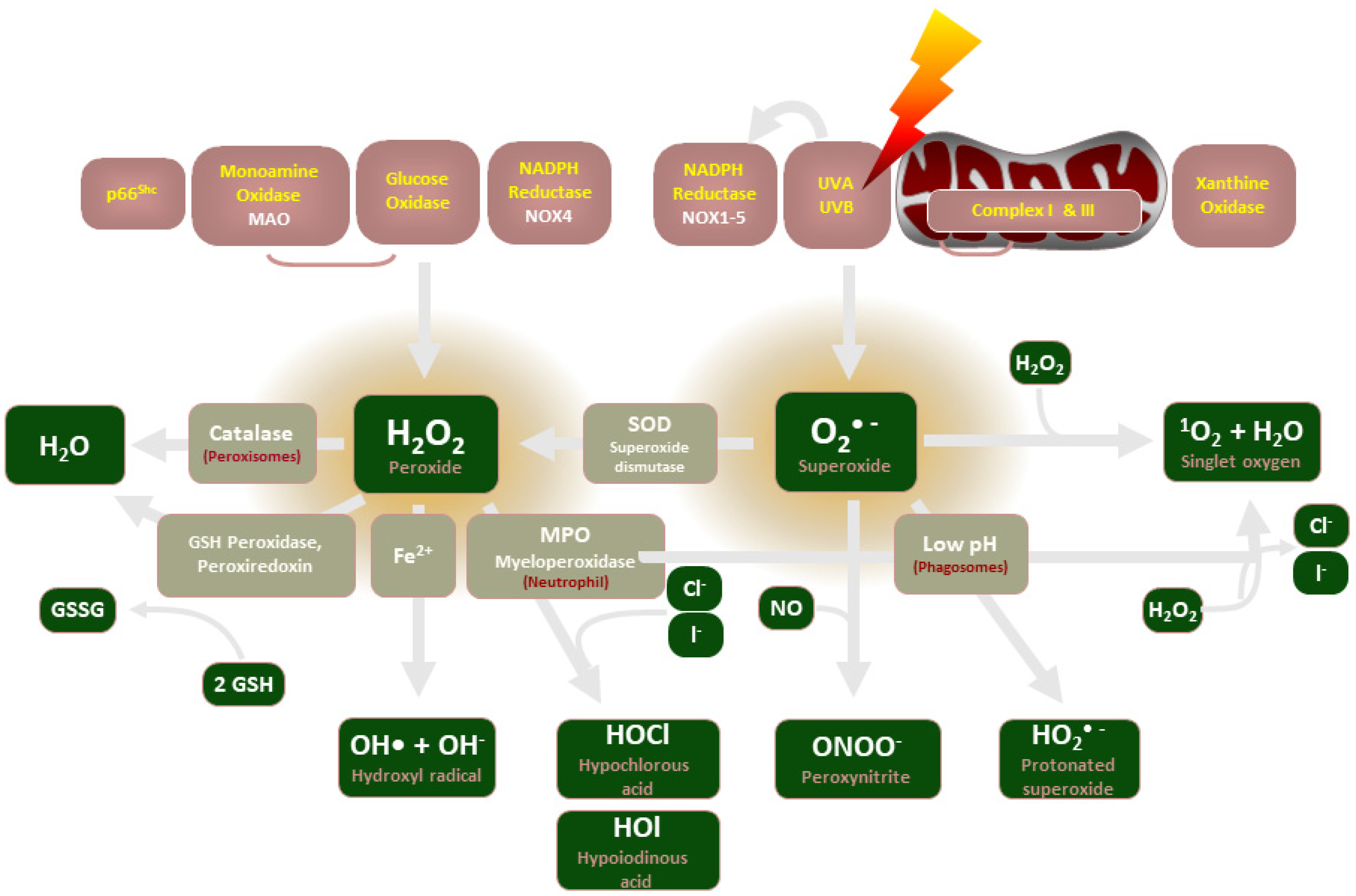
2. NADPH Oxidase Description
3. ROS and NADPH Oxidase in Recent Experimental Studies
4. NADPH Oxidases and ROS in Disease
5. NADPH Oxidase-Produced H2O2 Mediates Vascular Tone in Healthy Subjects during Exercise
6. RIRR, Endothelial Dysfunction and Angiogenesis
This entry is adapted from the peer-reviewed paper 10.3390/biom12060823
References
- Thompson, J.A.; Larion, S.; Mintz, J.D.; de Chantemèle, E.J.B.; Fulton, D.J.; Stepp, D.W. Genetic deletion of NADPH oxidase 1 rescues microvascular function in mice with metabolic disease. Circ. Res. 2017, 121, 502–511.
- Veith, C.; Boots, A.W.; Idris, M.; van Schooten, F.J.; van der Vliet, A. Redox Imbalance in Idiopathic Pulmonary Fibrosis: A Role for Oxidant Cross-Talk Between NADPH Oxidase Enzymes and Mitochondria. Antioxid. Redox Signal. 2019, 31, 1092–1115.
- Liang, S.; Kisseleva, T.; Brenner, D.A. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front. Physiol. 2016, 7, 17.
- Szanto, I.; Rubbia-Brandt, L.; Kiss, P.; Steger, K.; Banfi, B.; Kovari, E.; Herrmann, F.; Hadengue, A.; Krause, K.-H. Expression ofNOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathol. 2005, 207, 164–176.
- Juhasz, A.; Markel, S.; Gaur, S.; Liu, H.; Lu, J.; Jiang, G.; Wu, X.; Antony, S.; Wu, Y.; Melillo, G.; et al. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J. Biol. Chem. 2017, 292, 7866–7887.
- Lam, G.Y.; Huang, J.; Brumell, J.H. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin. Immunopathol. 2010, 32, 415–430.
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991.
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814.
- Brown, O.I.; Bridge, K.I.; Kearney, M.T. Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Glucose Homeostasis and Diabetes-Related Endothelial Cell Dysfunction. Cells 2021, 10, 2315.
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076.
- Helfinger, V.; Palfi, K.; Weigert, A.; Schröder, K. The NADPH Oxidase Nox4 Controls Macrophage Polarization in an NFκB-Dependent Manner. Oxidative Med. Cell. Longev. 2019, 2019, 3264858.
- Touyz, R.M.; Anagnostopoulou, A.; Rios, F.; Montezano, A.C.; Camargo, L.D.L. NOX5: Molecular biology and pathophysiology. Exp. Physiol. 2019, 104, 605–616.
- Petheő, G.L.; Kerekes, A.; Mihálffy, M.; Donkó, Á.; Bodrogi, L.; Skoda, G.; Baráth, M.; Hoffmann, O.I.; Szeles, Z.; Balázs, B.; et al. Disruption of the NOX5 Gene Aggravates Atherosclerosis in Rabbits. Circ. Res. 2021, 128, 1320–1322.
- Fusco, R.; Siracusa, R.; Gugliandolo, E.; Peritore, A.F.; D’Amico, R.; Cordaro, M.; Crupi, R.; Impellizzeri, D.; Gomiero, C.; Cuzzocrea, S.; et al. Micro Composite Palmitoylethanolamide/Rutin Reduces Vascular Injury through Modulation of the Nrf2/HO−1 and NF-kB Pathways. Curr. Med. Chem. 2021, 28, 6287–6302.
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and Cancer Metabolism. Clin. Cancer Res. 2012, 18, 5546–5553.
- Fry, E.A.; Inoue, K. c-MYB and DMTF1 in Cancer. Cancer Investig. 2019, 37, 46–65.
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure In Vivo. Arter. Thromb. Vasc. Biol. 2011, 31, 1368–1376.
- Muñoz, M.; Martínez, M.P.; López-Oliva, M.E.; Rodríguez, C.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernández, M.; Rivera, L.; Medina, J.S.; et al. Hydrogen peroxide derived from NADPH oxidase 4- and 2 contributes to the endothelium-dependent vasodilatation of intrarenal arteries. Redox Biol. 2018, 19, 92–104.
- Larsen, B.T.; Bubolz, A.H.; Mendoza, S.A.; Pritchard, K.A.; Gutterman, D.D. Bradykinin-Induced Dilation of Human Coronary Arterioles Requires NADPH Oxidase–Derived Reactive Oxygen Species. Arter. Thromb. Vasc. Biol. 2009, 29, 739–745.
- Xie, Y.; Nishijima, Y.; Zinkevich, N.S.; Korishettar, A.; Fang, J.; Mathison, A.J.; Zimmermann, M.T.; Wilcox, D.A.; Gutterman, D.D.; Shen, Y.; et al. NADPH oxidase 4 contributes to TRPV4-mediated endothelium-dependent vasodilation in human arterioles by regulating protein phosphorylation of TRPV4 channels. Basic Res. Cardiol. 2022, 117, 24.
- Wang, H.; Hartnett, M.E. Roles of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase in Angiogenesis: Isoform-Specific Effects. Antioxidants 2017, 6, 40.
- Damico, R.; Zulueta, J.J.; Hassoun, P.M. Pulmonary Endothelial Cell NOX. Am. J. Respir. Cell Mol. Biol. 2012, 47, 129–139.
- Meiners, S.; Eickelberg, O.; Königshoff, M. Hallmarks of the ageing lung. Eur. Respir. J. 2015, 45, 807–827.
- Nelkine, L.; Vrolijk, M.F.; Drent, M.; Bast, A. Role of antioxidants in the treatment of gastroesophageal reflux disease-associated idiopathic pulmonary fibrosis. Curr. Opin. Pulm. Med. 2020, 26, 363–371.
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2012, 1832, 1028–1040.
- Liu, B.; Zhang, X.; Zhang, F.-C.; Zong, J.-B.; Zhang, W.; Zhao, Y. Aberrant TGF-β1 signaling contributes to the development of primary biliary cirrhosis in murine model. World J. Gastroenterol. 2013, 19, 5828–5836.
- Bhardwaj, V.; Gokulan, R.C.; Horvat, A.; Yermalitskaya, L.; Korolkova, O.; Washington, K.M.; El-Rifai, W.; Dikalov, S.I.; Zaika, A.I. Activation of NADPH oxidases leads to DNA damage in esophageal cells. Sci. Rep. 2017, 7, 9956.
- Gole, H.; Tharp, D.L.; Bowles, D.K. Upregulation of Intermediate-Conductance Ca2+-Activated K+ Channels (KCNN4) in Porcine Coronary Smooth Muscle Requires NADPH Oxidase 5 (NOX5). PLoS ONE 2014, 2, e105337.
- Minnis, P.; Henry, K.; Keane, M.P. Reflux in idiopathic pulmonary fibrosis: Table 1. QJM Int. J. Med. 2015, 109, 7–10.
- Sukumar, P.; Viswambharan, H.; Imrie, H.; Cubbon, R.M.; Yuldasheva, N.; Gage, M.; Galloway, S.; Skromna, A.; Kandavelu, P.; Santos, C.X.; et al. Nox2 NADPH Oxidase Has a Critical Role in Insulin Resistance–Related Endothelial Cell Dysfunction. Diabetes 2013, 62, 2130–2134.
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119.
- Wilkinson-Berka, J.L.; Deliyanti, D.; Rana, I.; Miller, A.G.; Agrotis, A.; Armani, R.; Szyndralewiez, C.; Wingler, K.; Touyz, R.; Cooper, M.E.; et al. NADPH Oxidase, NOX1, Mediates Vascular Injury in Ischemic Retinopathy. Antioxid. Redox Signal. 2014, 20, 2726–2740.
- De Figueiredo, A.S.P.; Salmon, A.B.; Bruno, F.; Jimenez, F.; Martinez, H.G.; Halade, G.V.; Ahuja, S.S.; Clark, R.A.; DeFronzo, R.A.; Abboud, H.E.; et al. Nox2 mediates skeletal muscle insulin resistance induced by a high fat diet. J. Biol. Chem. 2015, 290, 13427–13439.
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913.
- Chrissobolis, S.; Banfi, B.; Sobey, C.G.; Faraci, F.M. Role of nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J. Appl. Physiol. 2012, 113, 184–191.
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 969.
- Durand, M.J.; Dharmashankar, K.; Bian, J.T.; Das, E.; Vidovich, M.; Gutterman, D.D.; Phillips, S.A. Acute exertion elicits a H2O2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension 2015, 65, 140.
- Radi, R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA 2018, 115, 5839–5848.
- Zinkevich, N.S.; Fancher, I.S.; Gutterman, D.D.; Phillips, S.A. Roles of NADPH oxidase and mitochondria in flow-induced vasodilation of human adipose arterioles: ROS-induced ROS release in coronary artery disease. Microcirculation 2017, 24, e12380.
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Circ. Physiol. 2011, 301, H647–H653.
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Physiol. 2017, 312, C749–C764.
- Fukai, T.; Ushio-Fukai, M. Cross-Talk between NADPH Oxidase and Mitochondria: Role in ROS Signaling and Angiogenesis. Cells 2020, 9, 1849.