 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Fengzhi Li | -- | 3046 | 2022-06-22 13:43:41 | | | |
| 2 | Beatrix Zheng | -129 word(s) | 2917 | 2022-06-23 10:34:48 | | | | |
| 3 | Beatrix Zheng | Meta information modification | 2917 | 2022-06-23 10:35:29 | | |
Video Upload Options
Molecular glue (MG) compounds are a type of unique small molecule that can change the protein–protein interactions (PPIs) and interactomes by degrading, stabilizing, or activating the target protein after their binging. These small-molecule MGs are gradually being recognized for their potential application in treating human diseases, including cancer. Evidence suggests that small-molecule MG compounds could essentially target any proteins, which play critical roles in human disease etiology, where many of these protein targets were previously considered undruggable. Intriguingly, most MG compounds with high efficacy for cancer treatment can glue on and control multiple key protein targets. On the other hand, a single key protein target can also be glued by multiple MG compounds with distinct chemical structures. The high flexibility of MG–protein interaction profiles provides rich soil for the growth and development of small-molecule MG compounds that can be used as molecular tools to assist in unraveling disease mechanisms, and they can also facilitate drug development for the treatment of human disease, especially human cancer.
1. CRBN-Involved CRL4 Complex-Mediated Degradation of MG-Targeted Proteins
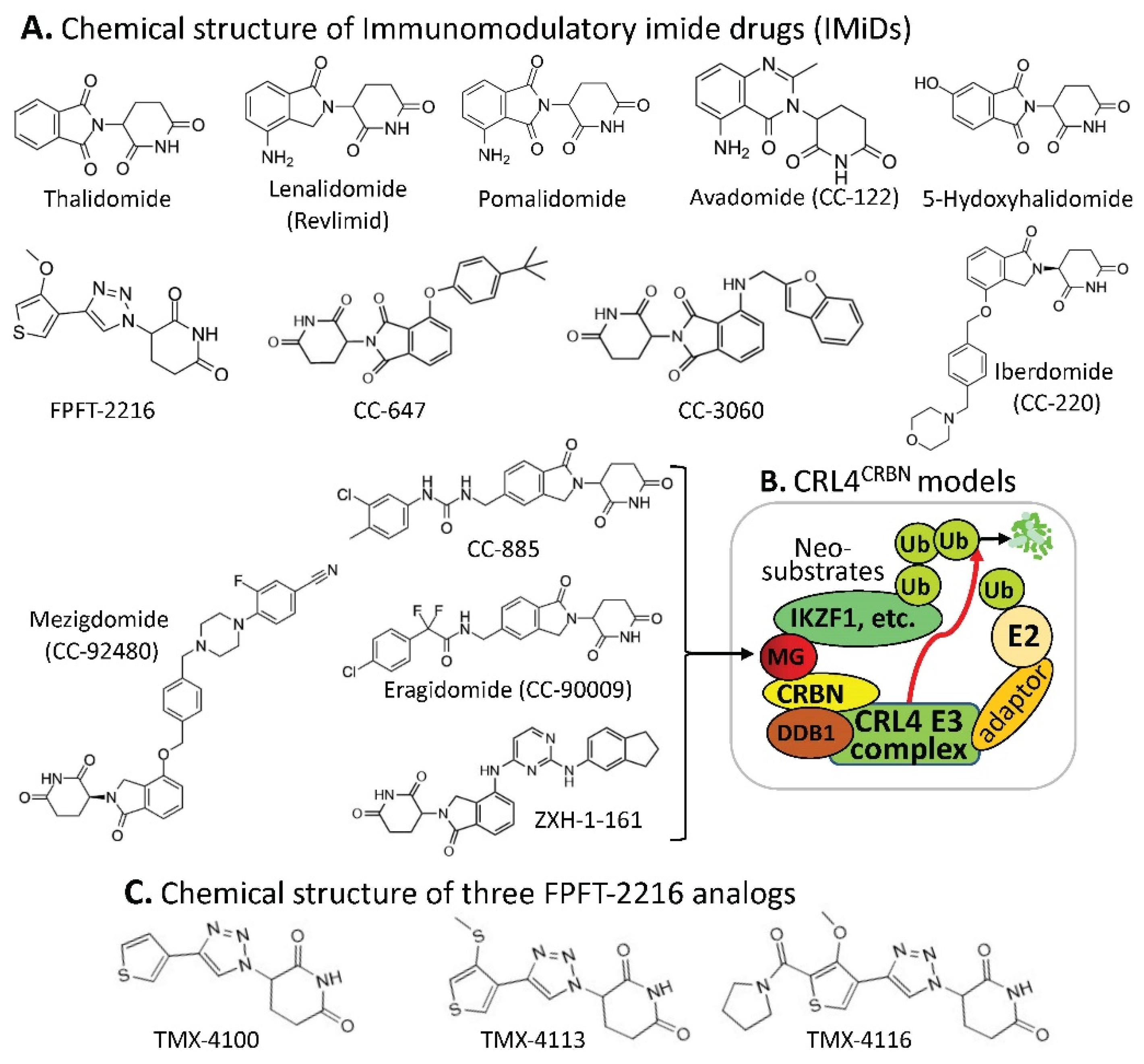
| MG Compounds | Neosubstrates/ Protein Targets |
Neosubstrates/ Protein Targets |
Molecular Glue (MG) Compounds |
|
|---|---|---|---|---|
| Thalidomide | IKZF1 #, IKZF3, ZNF692, ZNF276, SALL4, RNF166, ZBTB16, FAM83F, p63 | IKZF1 | thalidomide, lenalidomide, pomalidomide, avadomide/CC-122, FPFT-2216, iberdomide/CC-220, CC-3060, CC-92480, CC-885 | |
| Lenalidomide (Revlimid) |
IKZF1, IKZF3, ZFP91, ZFP692, ZNF276, ZNF653, ZNF827, SALL4, RNF166, WIZ1, CK1α, FAM83F, RAB28 | IKZF3 | thalidomide, lenalidomide, pomalidomide, avadomide/CC-122, iberdomide/CC-220, 92480, CC-885 | |
| Pomalidomide | IKZF1, IKZF3, ZFP91, ZFP692, ZNF276, ZNF653, ZNF827, SALL4, RNF166, GZF1, ZBTB39, ZNF98, WIZ1, ZBTB16, FAM83F, RAB28, DTWD1 | ZNF692 | thalidomide, lenalidomide, pomalidomide, | |
| Avadomide (CC-122) |
IKZF1, IKZF3, ZFP91 | ZNF276 | thalidomide, lenalidomide, pomalidomide, | |
| 5-hydroxy-thalidomide | SALL4, ZBTB16 | SALL4 | thalidomide, lenalidomide, pomalidomide, FPFT-2216, | |
| FPFT-2216 | IKZF1, CK1α | RNF166 | thalidomide, lenalidomide, pomalidomide, | |
| Iberdomide (CC-220) |
IKZF1, IKZF3, ZFP91, ZNF98 | ZBTB16 | thalidomide, pomalidomide, 5-hydroxythalidomide, CC-647, CC-3060 | |
| CC-647 | ZBTB16 | FAM83F | thalidomide, lenalidomide, pomalidomide, | |
| CC-3060 | ZBTB16, IKZF1, ZFP91, ZNF276 | p63 | thalidomide | |
| CC-92480 | IKZF1, IKZF3 | ZFP91 | lenalidomide, pomalidomide, avadomide/CC-122, iberdomide/CC-220, CC-3060 | |
| CC-885 | IKZF1, IKZF3, GSPT1, CK1α, PLK1, HBS1L | ZNF653 | lenalidomide, pomalidomide, | |
| CC-90009 | GSPT1 | ZNF827 | lenalidomide, pomalidomide, | |
| ZXH-1-161 | GSPT1, GSPT2 | WIZ1 | thalidomide, lenalidomide, | |
| CK1α | lenalidomide, FPFT-2216, CC-885 | |||
| RAB28 | lenalidomide, pomalidomide, | |||
| GZF1 | pomalidomide, | |||
| ZBTB39 | pomalidomide, | |||
| ZNF98 | pomalidomide, iberdomide/CC-220, | |||
| DTWD1 | pomalidomide, | |||
| ZNF276 | CC-3060 | |||
| GSPT1 | CC-885, CC-90009, ZXH-1-161 | |||
| PLK1 | CC-885 | |||
| HBS1L | CC-885 | |||
| GSPT2 | ZXH-1-161 |
2. DCAF15-Involved CRL4 Complex-Mediated Degradation of MG-Targeted Proteins
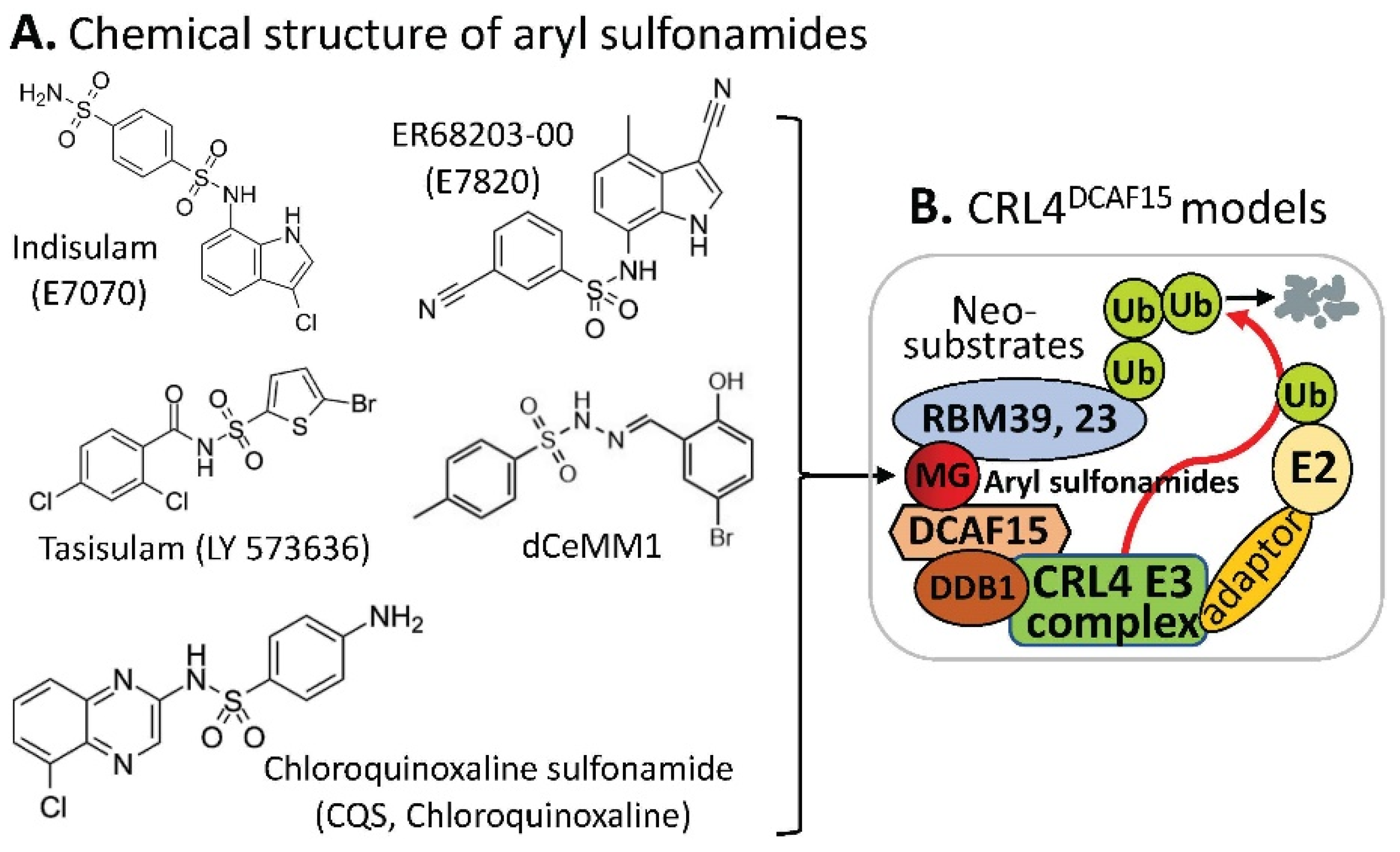
| Compounds | Protein Targets/Neosubstrates | Protein Targets | MG Compounds | |
|---|---|---|---|---|
| Indisulam | # RBM39, RBM23 | RBM39 | Indisulam, E7820, Tasisulam, CQS, dCeMM1 | |
| E7820 | RBM39, RBM23 | RBM23 | Indisulam, E7820, Tasisulam, CQS, | |
| Tasisulam | RBM39, RBM23 | |||
| CQS | RBM39, RBM23 | |||
| dCeMM1 | RBM39 |
3. Substrate Receptor-Independent E3 Ligase-Mediated Degradation of Cyclin K by MG
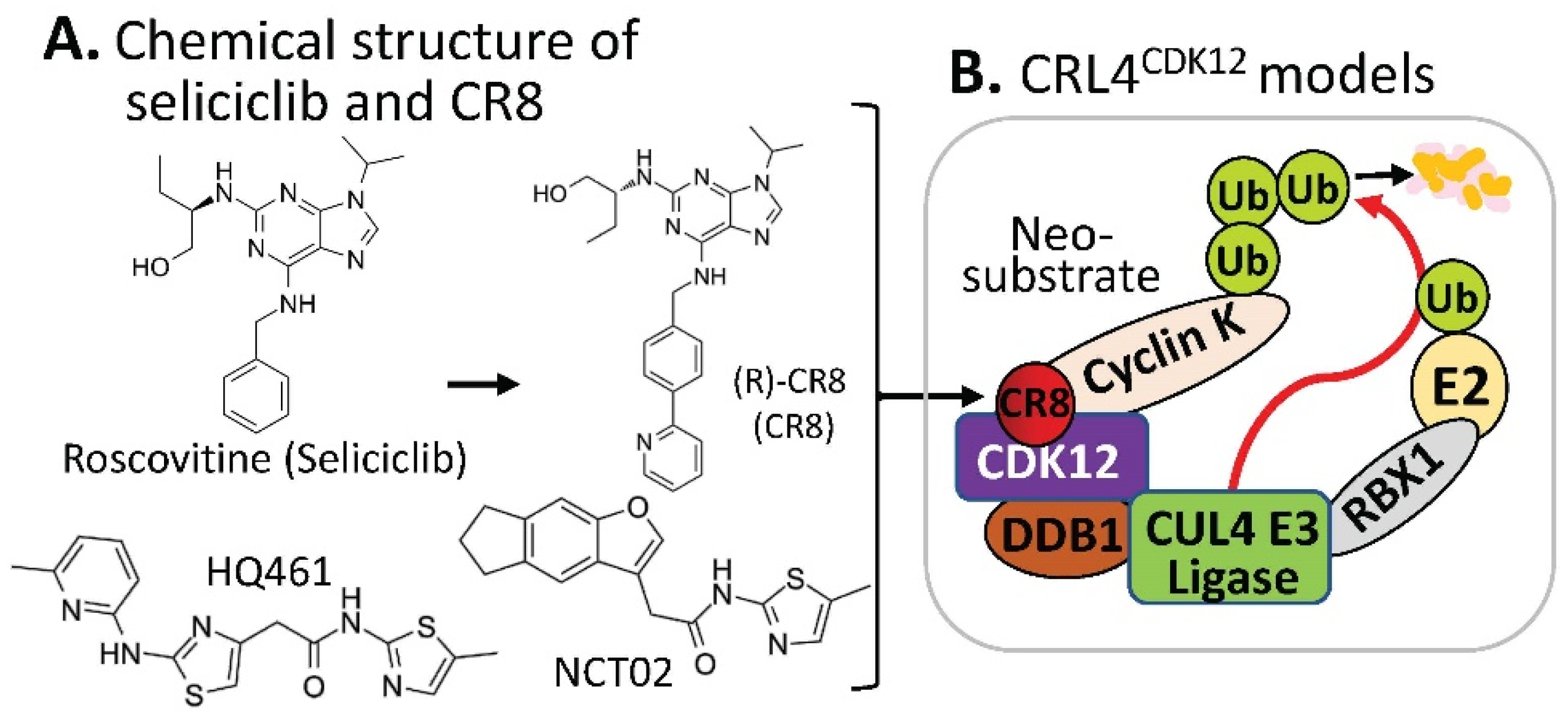
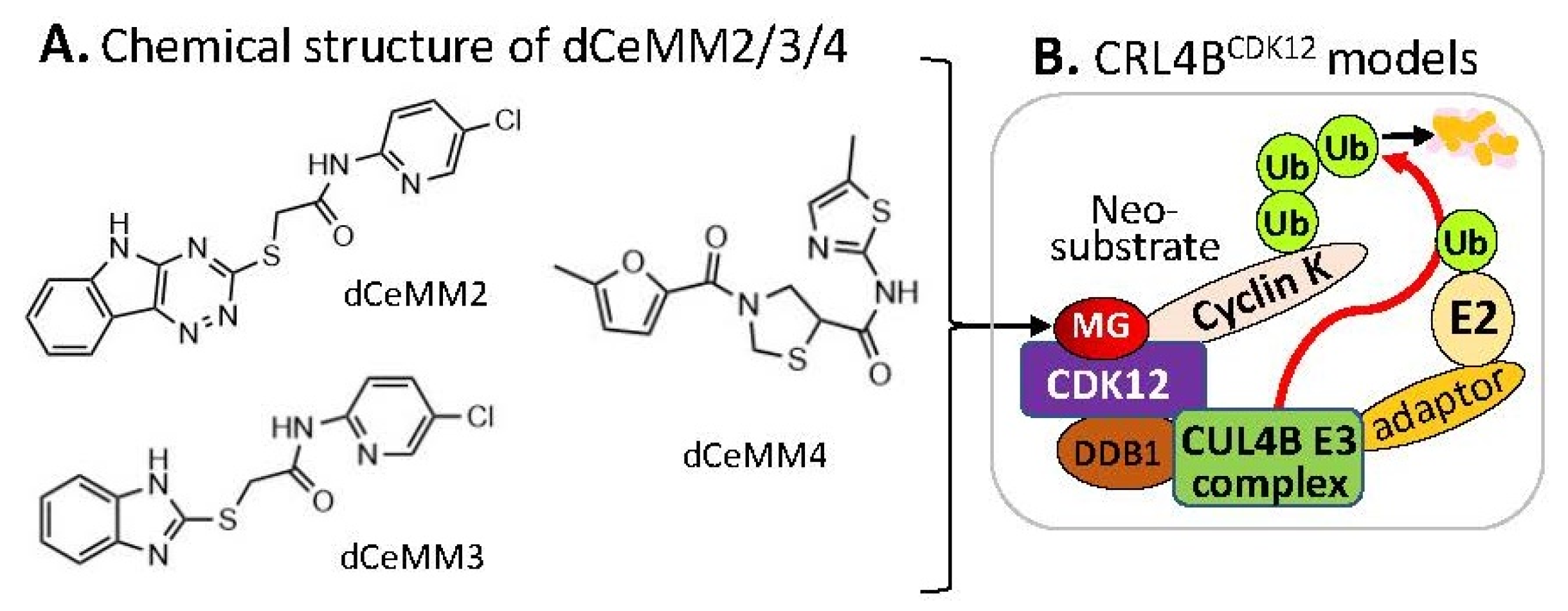
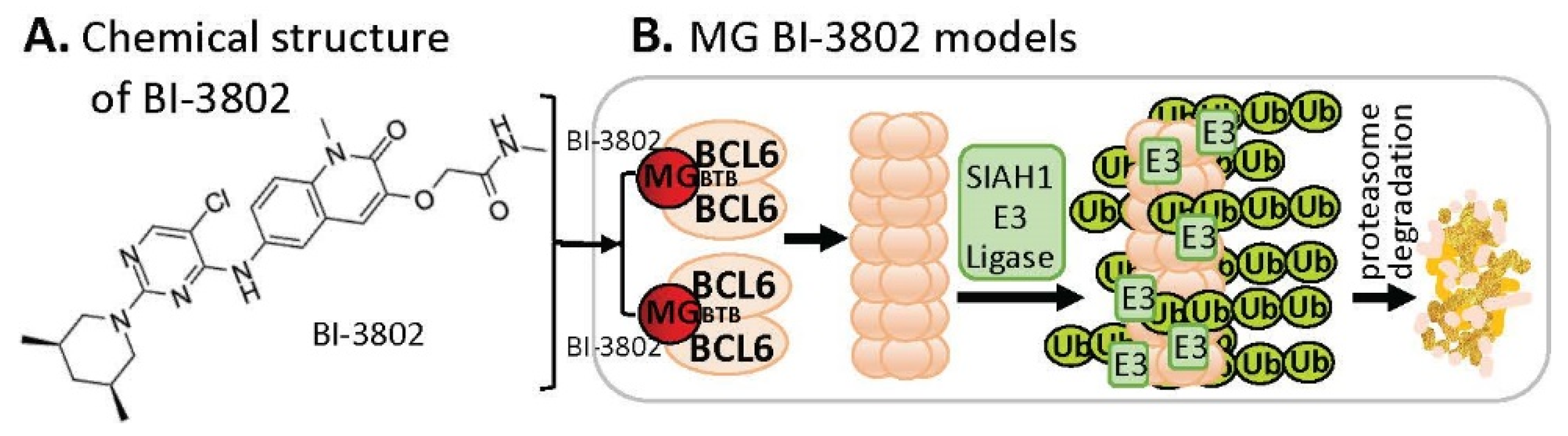
4. MG-Mediated Protein Homodimerization-Induced Protein Degradation
References
- Kozicka, Z.; Thoma, N.H. Haven’t got a glue: Protein surface variation for the design of molecular glue degraders. Cell Chem. Biol. 2021, 28, 1032–1047.
- Teng, M.; Lu, W.; Donovan, K.A.; Sun, J.; Krupnick, N.M.; Nowak, R.P.; Li, Y.D.; Sperling, A.S.; Zhang, T.; Ebert, B.L.; et al. Development of PDE6D and CK1alpha Degraders through Chemical Derivatization of FPFT-2216. J. Med. Chem. 2022, 65, 747–756.
- Chen, H.; Liu, J.; Kaniskan, H.U.; Wei, W.; Jin, J. Folate-Guided Protein Degradation by Immunomodulatory Imide Drug-Based Molecular Glues and Proteolysis Targeting Chimeras. J. Med. Chem. 2021, 64, 12273–12285.
- Pech, M.F.; Fong, L.E.; Villalta, J.E.; Chan, L.J.; Kharbanda, S.; O’Brien, J.J.; McAllister, F.E.; Firestone, A.J.; Jan, C.H.; Settleman, J. Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. Elife 2019, 8, e47362.
- Dong, G.; Ding, Y.; He, S.; Sheng, C. Molecular Glues for Targeted Protein Degradation: From Serendipity to Rational Discovery. J. Med. Chem. 2021, 64, 10606–10620.
- Singh, S.; Quarni, W.; Goralski, M.; Wan, S.; Jin, H.; Van de Velde, L.A.; Fang, J.; Wu, Q.; Abu-Zaid, A.; Wang, T.; et al. Targeting the spliceosome through RBM39 degradation results in exceptional responses in high-risk neuroblastoma models. Sci. Adv. 2021, 7, eabj5405.
- Nijhuis, A.; Sikka, A.; Yogev, O.; Herendi, L.; Balcells, C.; Ma, Y.; Poon, E.; Eckold, C.; Valbuena, G.N.; Xu, Y.; et al. Indisulam targets RNA splicing and metabolism to serve as a therapeutic strategy for high-risk neuroblastoma. Nat. Commun. 2022, 13, 1380.
- Gosavi, P.M.; Ngan, K.C.; Yeo, M.J.R.; Su, C.; Li, J.; Lue, N.Z.; Hoenig, S.M.; Liau, B.B. Profiling the Landscape of Drug Resistance Mutations in Neosubstrates to Molecular Glue Degraders. ACS Cent. Sci. 2022, 8, 417–429.
- Benson, C.; White, J.; De Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37.
- Oumata, N.; Bettayeb, K.; Ferandin, Y.; Demange, L.; Lopez-Giral, A.; Goddard, M.L.; Myrianthopoulos, V.; Mikros, E.; Flajolet, M.; Greengard, P.; et al. Roscovitine-derived, dual-specificity inhibitors of cyclin-dependent kinases and casein kinases 1. J. Med. Chem. 2008, 51, 5229–5242.
- Bettayeb, K.; Oumata, N.; Echalier, A.; Ferandin, Y.; Endicott, J.A.; Galons, H.; Meijer, L. CR8, a potent and selective, roscovitine-derived inhibitor of cyclin-dependent kinases. Oncogene 2008, 27, 5797–5807.
- Yu, C.; Mannan, A.M.; Yvone, G.M.; Ross, K.N.; Zhang, Y.L.; Marton, M.A.; Taylor, B.R.; Crenshaw, A.; Gould, J.Z.; Tamayo, P.; et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat. Biotechnol. 2016, 34, 419–423.
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anti-cancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248.
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508.
- Slabicki, M.; Kozicka, Z.; Petzold, G.; Li, Y.D.; Manojkumar, M.; Bunker, R.D.; Donovan, K.A.; Sievers, Q.L.; Koeppel, J.; Suchyta, D.; et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 2020, 585, 293–297.
- Mayor-Ruiz, C.; Bauer, S.; Brand, M.; Kozicka, Z.; Siklos, M.; Imrichova, H.; Kaltheuner, I.H.; Hahn, E.; Seiler, K.; Koren, A.; et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat. Chem. Biol. 2020, 16, 1199–1207.
- Lv, L.; Chen, P.; Cao, L.; Li, Y.; Zeng, Z.; Cui, Y.; Wu, Q.; Li, J.; Wang, J.H.; Dong, M.Q.; et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger cyclin K degradation. eLife 2020, 9, e59994.
- Dieter, S.M.; Siegl, C.; Codo, P.L.; Huerta, M.; Ostermann-Parucha, A.L.; Schulz, E.; Zowada, M.K.; Martin, S.; Laaber, K.; Nowrouzi, A.; et al. Degradation of CCNK/CDK12 is a druggable vulnerability of colorectal cancer. Cell Rep. 2021, 36, 109394.
- Kerres, N.; Steurer, S.; Schlager, S.; Bader, G.; Berger, H.; Caligiuri, M.; Dank, C.; Engen, J.R.; Ettmayer, P.; Fischerauer, B.; et al. Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep. 2017, 20, 2860–2875.
- Slabicki, M.; Yoon, H.; Koeppel, J.; Nitsch, L.; Roy Burman, S.S.; Di Genua, C.; Donovan, K.A.; Sperling, A.S.; Hunkeler, M.; Tsai, J.M.; et al. Small-molecule-induced polymerization triggers degradation of BCL6. Nature 2020, 588, 164–168.


