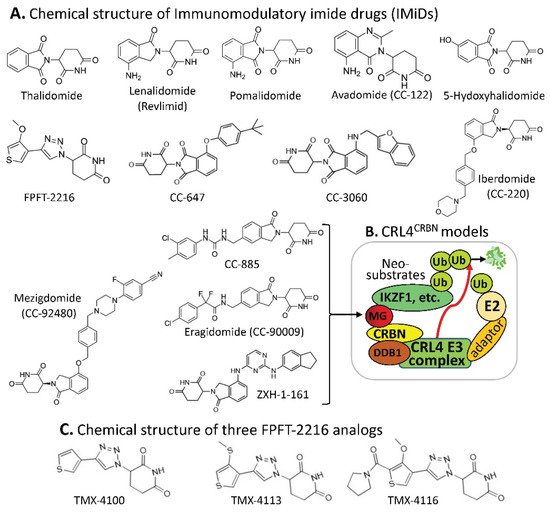
1. CRBN-Involved CRL4 Complex-Mediated Degradation of MG-Targeted Proteins
Human CRBN (cereblon) was originally identified as a candidate protein factor for an autosomal recessive form of mild mental retardation. Relevant to the ubiquitination and degradation of MG-targeted proteins, CRBN is the substrate receptor of the CRL4 (cullin 4-RING E3 ligase) complex, which may also include damaged DNA binding protein 1 (DDB1), among others (Figure 1B). Based on the studies documented in the literature, particular protein members in the CRL4 complex depend on bothboth the property of specific MG compounds and specific, the property of specific MG compounds and specific, targeted proteins for ubiquitination (examples are shown later). This CRL4CRBN complex through CRBN can be engaged in ubiquitinating many types of proteins for proteasome degradation.
Figure 1. (A) The IMiD-type MG compounds’ chemical structures; (B) the substrate receptor CRBN-involved E3 ligase protein complex model. Each compound in (A) was documented to engage the same E3 ligase protein complex (CRL4CRBN) to polyubiquitinate the compound’s neosubstrates shown in Table 1. Then, the polyubiquitinated neosubstrates/protein targets in Table 1 would be degraded through the ubiquitination proteasome pathway; and (C) chemical structure of FPJFT-2216-derived three new small molecules (TMX-4100, TMX-4113, TMX-4116).
It has been reported that the CRL4
CRBN complex E3 ligase can be used by the small-molecule MG of thalidomide and its structural analogs (lenalidomide/Revlimid, pomalidomide, avadomide, 5-hydroxythalidomide, FPFT-2216, CC-647, CC-3060, iberdomide, mezigdomide, CC-885, eragidomide, and ZXH-1-161) to ubiquitinate the MG-recruited/bound protein neosubstrates (
Table 1), and these have been well-reviewed recently
[1][12] (
Figure 1A,B). However, based on the literature-documented studies, one interesting phenomenon that may be worthy of emphasis is that thalidomide and its individual analogs with different chemical structures can share the same CRL4
CRBN complex E3 ligase for ubiquitinating different protein neosubstrates glued by them. Specifically, a single MG compound can glue/bind to multiple different neosubstrate protein targets for CRL4
CRBN ubiquitination, while the same protein target/neosubstrate can also be glued by thalidomide and multiple thalidomide analogs (
Table 1).
We anticipate that neosubstrate protein targets for these MG compounds will be found in the future. For example, Teng et al. recently demonstrated that FPFT-2216 (
Figure 1A) and its chemically modified analogs TMX-4100 and TMX-4113 but not TMX-4116 (
Figure 1C) could selectively degrade PDE6D (Phosphodiesterase 6D)
[2][13] through a similar approach as shown in
Figure 1B.
The observation summarized in Table 1 has revealed an important concept for further exploration in the research community. That is, as long as the neosubstrate protein targets bound by an MG compound have good specificity, multiple protein targets that can be glued by a single compound may not be an issue with regard to potential toxicity in normal tissues. A good example of this notion is that lenalidomide (Revlimid, Figure 1A) can glue at least 13 neosubstrate protein molecules (Table 1) to be ubiquitinated by CRL4CRBN for proteasome degradation (Figure 1A,B). Revlimid has been successfully commercialized without serious toxicity issues that could block its approval by the FDA. In fact, Revlimid has become one of the most successful anticancer drugs for the treatment of human multiple myeloma (MM) in the clinic. In this regard, it is likely that small-molecule MG compounds that can specifically glue multiple oncogenic proteins for degradation and/or inhibition of their oncogenic activity may have greater potential to be developed into an anticancer drug. Based on previously documented examples, most anticancer drugs that failed in the later phase II and phase III clinical trials were due to their insufficient antitumor efficacy but not due to their toxicity. This is consistent with the fact that most anticancer drug phase I clinical trials were successful, and researchers who performed the studies often concluded that the drug is well-tolerated and warranted further phase II clinical studies.
(The following part in Section 1 together with the figure 2 can be removed without any issue if you want this article to be succinct. Otherwise, leave it as it is.)
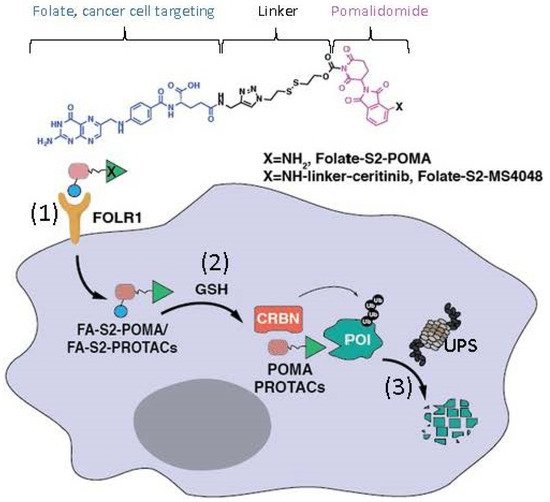
Additionally, Chen et al. recently reported a modified MG model derived from a pomalidomide-based compound
[3][14]. Specifically, these authors linked a folate group to the MG pomalidomide or the pomalidomide-based PROTAC as prodrugs to specifically recognize the folate receptor α (FOLR1)-positive cancer cells
(Figure 2). These authors demonstrated that such prodrugs target FOLR1-positive cancer cells and release the folate-tethered MG/PROTAC, which then degrade the protein targets in FOLR1-positive cancer cells in a CRBN- and proteasome-dependent manner, but this did not occur in FOLR1-negative cancer cells
[3][14]. This is an interesting drug design that extends
the our
esearchers' views on MG/PROTAC drugs to more specifically target the target-/bait-expressed cancer cells but not target-/bait-negative normal cells by using an additional cancer cell surface target as a bait. However,
the researchers swe should always keep in mind that if an MG/PROTAC drug’s cellular protein target/bait can express in both bait-positive and bait-negative cancer cells, an MG/PROTAC drug designed in this way would not be able to degrade their cellular protein targets in the bait-negative cancer cells. In short, additional considerations are necessary when designing such prodrugs for cancer treatment.
Figure 2. This diagram shows the mode of action of folate–pomalidomide and folate-conjugated pomalidomide-based PROTACs. Upon binding FOLR1 on the cell membrane (1), folate-pomalidomide or folate-conjugated IMiD-based PROTACs are transported into cells, and the active pomalidomide or PROTACs are released after the reduction by endogenous GSH (2). The active pomalidomide or PROTACs recruit endogenous CRBN E3 ligase (similar to the model shown in Figure 1B), leading to polyubiquitination and subsequent degradation of the glued proteins of interest (POIs, neosubstrates) by the UPS (3). This figure is adapted from an original paper by Chen et al. [14].
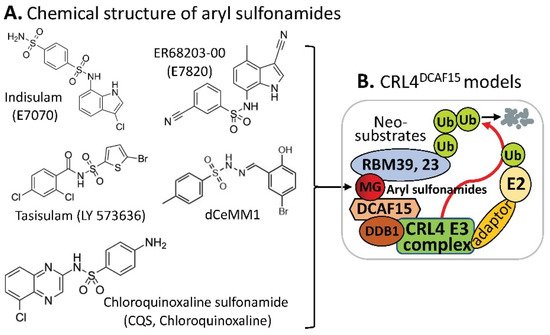
2. DCAF15-Involved CRL4 Complex-Mediated Degradation of MG-Targeted Proteins
Human DDB1 and Cullin 4-associated factor 15 (DCAF15) protein is encoded by the
DCAF15 gene, and a recent study revealed that genetic disruption of DCAF15 strongly sensitized cancer cells to natural killer (NK)-mediated clearance
[4][15]. Similar to the case of the CRL4
CRBN complex E3 ligase in which CRBN acts as a substrate receptor of CRL4, DCAF15 can also be a part of the CRL4 complex E3 ligase, acting as the CRL4 E3 ligase complex’s substrate receptor/adaptor to be glued on protein neosubstrates, RBM39 and/or RBM23 through the MG compounds, aryl sulfonamides (e.g., indisulam, E7820, tasisulam, dCeMM1, CQS, etc.) for RBM39 and/or RBM23 to be ubiquitinated for proteasome degradation (
Figure 23). The details in these studies have been recently reviewed
[1][5][9,12].
Figure 23. (A) The aryl sulfonamide type MG compounds’ chemical structures; (B) the substrate receptor DCAF15-involved E3 ligase protein complex model. Each compound in (A) was documented to engage the same E3 ligase protein complex (CRL4DCAF15) to polyubiquitinate the compound’s neosubstrates shown in Table 2. Then, the polyubiquitinated neosubstrates/protein targets in Table 2 would be degraded through the ubiquitination proteasome pathway.
Similar to the observation revealed from the data summarized in
Table 1, the CRL4
DCAF15-based MG aryl sulfonamides can share protein neosubstrates and vice versa; a protein substrate can share MG aryl sulfonamides
[1][12] (
Table 2).
Recently, two papers reported that the RNA splicing regulator RBM39 is important for Myc-driven neuroblastoma survival
[6][7][16,17]. The indisulam-mediated degradation of RBM39 needs the presence of a high-level DCAF15 expression and is highly efficacious against neuroblastoma, leading to significant responses in multiple high-risk disease models, without overt toxicity
[6][16]. Furthermore, in neuroblastoma models, indisulam induces rapid loss of RBM39, accumulation of splicing errors, and the growth inhibition in a DCAF15-dependent manner with metabolome perturbations as well as the mitochondrial dysfunction
[7][17]. Remarkably, complete tumor regression was observed in xenograft models and the Th-MYCN transgenic model of neuroblastoma after intravenous indisulam treatment daily at a dose of 25 mg/kg for 8 days (xenograft mouse model) or for 7 days (Th-MYCN mouse model)
[7][17]. These authors indicated that the dose of 25 mg/kg used in the studies is equivalent to the clinically tolerated dose range of 500–700 mg/m
2 reported in adult humans
[7][17]. Meanwhile, these authors further confirmed RBM39 loss, RNA splicing, and metabolic changes in vivo after indisulam treatment
[7][17].
Most recently, just before submitting this article for publication, Gosavi et al. reported their interesting findings on the neosubstrates GSPT1 and RBM39 mutation-induced MG drug resistance
[8][18]. Specifically, these authors used the CRISPR-suppressor scanning approach and identified mechanistic classes of CC-885 and ZXH-1-161 resistance mutations in GSPT1, as well as E7820 and Indisulam resistance mutations in RBM39; the mutations in GSPT1 and RBM39 are resistant to the CRBN- (
Figure 1) and DCAF15 (
Figure 23)-involved E3 ligase-mediated GSPT1 and RBM39 ubiquitination for degradation
[8][18]. These authors found that, while many mutations directly alter the ternary complex heterodimerization surface, distal resistance sites were also identified. CC-885 resistance mutations altered the GSPT1 β-hairpin structural degron and impaired GSPT1 degradation, while E7820 resistance mutations in different domains of RBM39 were operated via distinct mechanisms. The studies further indicated that mutations distal to the RBM39 RRM2 helix 1 structural degron altered maximum levels of RBM39 degradation to abrogate E7820 cytotoxicity, and that resistance mutation sites across targeted protein degradation targets exhibit low levels of sequence conservation
[8][18]. Together, these authors summarized that their study identifies common resistance mechanisms for MG degraders and outlines a general approach to survey neosubstrate requirements necessary for effective degradation
[8][18].
3. Substrate Receptor-Independent E3 Ligase-Mediated Degradation of Cyclin K by MG
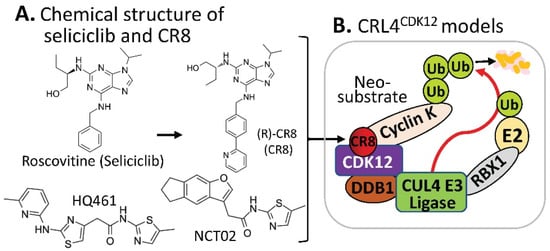
The CDK inhibitor, purine (R) roscovitine (also called CYC202 or Seliciclib) had been used in some clinical trials
[9][19], and recently, a phase II clinical trial with cystic fibrosis patients was completed, which has been reported on the “clinicaltrials.gov” website at
https://clinicaltrials.gov/ct2how/NCT02649751. Based on the (R)-roscovitine chemical structure, Ounata et al. synthesized a series of (R)-roscovitine analogs including a compound called (R)-CR8 (
Figure 34A)
[10][20]. Further studies indicated that (R)-CR8 selectively inhibits CDK1/2/3/5/7/9 among the 108 kinases tested and is more potent than (R)-roscovitine at inhibiting these kinases and at inducing apoptotic cell death parameters (MTS reduction, 40-fold; lactate dehydrogenase release, 35-fold; caspases activation, 68-fold; and PARP cleavage, 50-fold). Generally speaking, (R)-roscovitine works at a low µM level, while (R)-CR8 works at a low nM level. Convincingly, the improved cell death-inducing activity of (R)-CR8 over (R)-roscovitine was observed in 25 different cancer cell lines including colon (HCT116, LS174T), hepatoma (Huh7, F1) and neuroblastoma (SH-SY5Y)
[11][21].
Figure 34. (A) The chemical structure of roscovitine/Seliciclib, CR8, HQ461, and NCT02 as MG compounds; (B) the substrate receptor-independent E3 ligase protein complex model for ubiquitination of the neosubstrate cyclin K. The MG compound CR8 shown in (A) was found through a substrate receptor-independent manner (neither CRBN nor DCAF15 being involved) to glue CDK12–cyclin K directly on DDB1–CUL4 E3 ligase complex to polyubiquitinate cyclin K. Then, the polyubiquitinated cyclin K would be degraded through the ubiquitination proteasome pathway. HQ461 and NCT02 may use a mechanism similar to CR8.
Intriguingly, by using a molecular barcoding method called PRISM that was previously developed with comprehensive resources (compounds and cell lines)
[12][13][14][22,23,24], and through systematically mining databases for correlations between the cytotoxicity of 4518 clinical and preclinical small molecules and the expression levels of E3 ligase components across hundreds of human cancer cell lines, Stabicki et al. identified (R)-CR8 (CR8) as a molecular glue degrader compound to deplete cyclin K
[15][25]. These authors found that CR8-induced degradation of cyclin K is dependent on DDB1 and CDK12
[15][25]. Specifically, CR8 binds to the CDK12 ATP pocket site to form CDK12-CR8; then the CDK-bound form of CR8 has a solvent-exposed pyridyl moiety that induces the formation of a complex between CDK12-cyclin K and the CUL4 adaptor proteins, DDB1 and RBX1 (RING-box protein 1), bypassing the requirement of a canonical substrate receptor (e.g., not requiring CRBN or DCAF15) and presenting cyclin K for ubiquitination and degradation (
Figure 34B). In other words, CR8-engaged CDK12–cyclin K is recruited to the CUL4–RBX1–DDB1 ligase core through DDB1, and CR8 tightens the complex sufficiently to drive CR8-induced ubiquitination and then degradation of cyclin K in the absence of a canonical substrate receptor
[15][25] (
Figure 34B). These authors believe that their studies demonstrated that chemical alteration of surface-exposed moieties can confer gain-of-function glue properties to an inhibitor
[15][25].
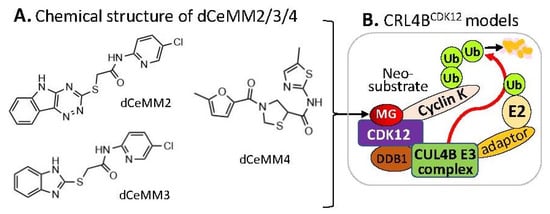
Alternatively, by using a different approach/strategy based on a 2000 cytostatic/cytotoxic small-molecules (PLACEBO in-house collection) screening in hyponeddylated cells coupled to a multi-omics (proteomics, RNA-Seq, etc.) target deconvolution campaign, Mayor-Ruiz et al. found several chemical structural distinct compounds (dCeMM2, dCeMM3, dCeMM4,
Figure 45A) acting as MG degraders that could also induce cyclin K degradation in a low µM level
[16][26]. Their studies indicated that dCeMM2/3/4 compounds-induced cyclin K degradation is mediated via a CRL4B ligase complex without the need for a substrate receptor (e.g., CRBN or DCAF15,
Figure 45B), and thus functionally segregating this mechanism from MG degraders that need a canonical substrate receptor (SP)
[16][26]. More details were recently reviewed by Dong et al.
[5][9].
Figure 45. (A) The chemical structure of dCeMM2, dCeMM3, and dCeMM4 as MG compounds; (B) the substrate receptor-independent E3 ligase protein complex model for ubiquitination of the neosubstrate cyclin K. The MG compounds dCeMM2/3/4 shown in (A) was found through a substrate receptor-independent manner (neither CRBN nor DCAF15 being involved) to glue CDK12-cyclin K directly on DDB1–CUL4B E3 ligase complex to polyubiquitinate cyclin K. Then, the polyubiquitinated cyclin K would be degraded through the ubiquitination proteasome pathway.
Furthermore, by employing a loss-of-function and gain-of-function genetic screening in human cancer cells, followed by biochemical reconstitution, Lv et.al found a new MG (named HQ461,
Figure 34A) that could degrade cyclin K
[17][27] in a mechanism very similar to that of CR8
[15][25] (
Figure 34B). More details were recently reviewed
[5][9].
Interestingly, by screening a small-molecule library against primary colorectal cancer (CRC) heterogeneous spheroids, Dieter et al. found a compound named NCT02 with a chemical structure similar to HQ461 (
Figure 4A) that acts as an MG degrader to induce ubiquitination of cyclin K (CCNK) and proteasomal degradation of both CCNK and its partner CDK12
[18][28].
4. MG-Mediated Protein Homodimerization-Induced Protein Degradation
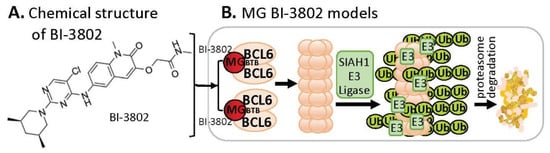
B-cell lymphoma 6 (BCL6) is known to be an oncogenic transcriptional factor. High-throughput screening (HTS) with a library of ~1.7 M compounds, followed by carrying out various narrowing-down and alternative confirmation approaches, led to the discovery of a BCL6 MG small-molecule degrader named BI-3802
[19][29] (
Figure 56A). In contrast to the MG compound models that MGs directly glue both their protein neosubstrates and the E3 ligase complex (very similar to PRTOTAC without the linker structure) to induce the glued protein to be polyubiquitinated for proteasome degradation, studies with BI-3802 revealed that the BCL6 monomer protein molecule can be dimerized by the small-molecule MG BI-3802 through the broad-complex, tramtrack, and bric-a-brac (BTB) domain of BCL6 and then assembled into BCL6 filaments, which then facilitates to induce BCL6 ubiquitination by E3 ligase SIAH1 for BCL6 degradation via the proteasome pathway
[20][30] (
Figure 56B).
Figure 56. (A) The chemical structure of BI-3802 as a special MG compound; (B) the diagram model for BI-3802-induced BCL6 polymerization, ubiquitination, and degradation. BI-3802 through the BTB domain of BCL6 makes BCL6 dimerization and then induces the dimerized BCL6 formation of helical filament. The polymerized BCL6 enhances the E3 ligase SIAHI interaction and ubiquitination of the polymerized BCL6 protein for degradation through the ubiquitination proteasome pathway.