Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yoshihiko Hara | -- | 2440 | 2022-06-21 06:49:02 | | | |
| 2 | Conner Chen | Meta information modification | 2440 | 2022-06-21 08:37:32 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Usuda, Y.; Nishio, Y.; Nonaka, G.; Hara, Y. Microbial Production Potential of Pantoea ananatis. Encyclopedia. Available online: https://encyclopedia.pub/entry/24247 (accessed on 24 July 2026).
Usuda Y, Nishio Y, Nonaka G, Hara Y. Microbial Production Potential of Pantoea ananatis. Encyclopedia. Available at: https://encyclopedia.pub/entry/24247. Accessed July 24, 2026.
Usuda, Yoshihiro, Yousuke Nishio, Gen Nonaka, Yoshihiko Hara. "Microbial Production Potential of Pantoea ananatis" Encyclopedia, https://encyclopedia.pub/entry/24247 (accessed July 24, 2026).
Usuda, Y., Nishio, Y., Nonaka, G., & Hara, Y. (2022, June 21). Microbial Production Potential of Pantoea ananatis. In Encyclopedia. https://encyclopedia.pub/entry/24247
Usuda, Yoshihiro, et al. "Microbial Production Potential of Pantoea ananatis." Encyclopedia. Web. 21 June, 2022.
Copy Citation
Pantoea ananatis, a gram-negative bacterium belonging to the Erwiniaceae family, is a well-known phytopathogen isolated from many ecological niches and plant hosts. However, this bacterium also provides us with various beneficial characteristics, such as the growth promotion of their host plants and increased crop yield.
Pantoea ananatis
microbial production
1. Introduction
Pantoea ananatis is a gram-negative, rod-shaped, aerobic, or facultatively anaerobic bacteria belonging to the class Gammaproteobacteria and was recently reclassified into the family Erwiniaceae from Enterobacteriaceae [1][2]. P. ananatis was first described as Erwinia ananas by Serrano [3]. Some strains of Enterobacter agglomerans, Erwinia herbicola, and Erwinia milletiae that form part of the E. herbicola–E. agglomerans complex was assigned to the genus Pantoea [4]. Later, Pantoea uredovora, a pathogen of Puccinia graminis, was shown to have a high level of genomic relatedness to P. ananas, and the two species were synonymized [5]. P. ananas proposed by Mergaert et al. [5] was corrected to P. ananatis by Trüper and De’Clari [6]. Due to this phylogenetic history, P. ananatis contains various kinds of plant pathogenic strains [1]. At the same time, strains that promote plant growth that are applicable to bioremediation, or have lignocellulose degradation capacity, have been found in recent years [7].
In the field of microbial biomanufacturing, Escherichia coli, a model organism and a member of the family Enterobacteriaceae, which is a gram-negative facultative anaerobic rod, has been used industrially for the production of various substances because of its high growth rate and sugar consumption activity in the neutral pH range [8][9]. Corynebacterium glutamicum, a gram-positive, rod-shaped bacterium, is capable of L-glutamate fermentation [10] and has been used for the industrial production of many substances by taking advantage of its characteristic cell surface [11][12]. To date, bacterial fermentation production has been realized by mainly using E. coli and C. glutamicum, and many tools for genetic engineering have been developed for both strains [9]. However, it was clear that if both strains had the trait of robustness to pH, they would be more desirable industrial substance-producing bacteria. P. ananatis AJ13355 was isolated as an acidophilic bacterium [13] and has been used as an excellent host organism in previous studies to produce amino acids such as L-glutamate [14], L-cysteine [15], and isoprenoids [16][17]. The emergence of strains closely related to E. coli and advantageous for industrial production is important in terms of increasing the potential for substance production.
2. Characteristic of Pantoea ananatis
P. ananatis has been isolated from various environments and hosts and is well-known for its phytopathogenicity. P. ananatis causes disease symptoms in many economically important agronomic crops and forest tree species worldwide [1]. However, several strains have been known to improve the growth of plants [7], such as papaya [18], red pepper [19], sugarcane [20][21], poplar [22], and rice [23][24]. Kim et al. [19][25] determined the genome sequence of P. ananatis B1–9 isolated from the rhizosphere of the green onion and reported that enhanced red pepper crop yield by approximately three times and showed phosphate solubilization, sulfur oxidation, nitrogen fixation, and indole-3-acetic acid (IAA) production activities. P. ananatis AMG521, isolated as an endophyte from rice paddies, showed the capacity to synthesize siderophores, cellulose, and IAA, and the capacity to solubilize phosphate and increase rice yield [23]. P. ananatis strain 1.38, isolated from the rhizosphere of rice (Oryza sativa L.), has been reported to have phosphate solubilization activity, siderophore and auxin production, and cellulose, lipase, and pectinase activities [24]. It has long been known that P. ananatis produces carotenoids, and it has been reported that introducing the carotenoid biosynthesis genes of P. ananatis into a microorganism that does not produce carotenoids leads to the production of lycopene, astaxanthin, and β-carotene [26][27]. Ten complete genomes of P. ananatis have been determined and registered. Four were pathogenic and four had useful traits (Table 1).
Table 1. Complete genome of Pantoea ananatis.
| Strain | Assembly | Chromosome Size | CDS | Plasmids | Classification | Source | Reference |
|---|---|---|---|---|---|---|---|
| Pantoea ananatis LMG 20103 | GCA_000025405.2 | 4,703,373 | 4272 | 0 | Plant pathogen | Eucalyptus | [28] |
| P. ananatis AJ13355 | GCA_000270125.2 | 4,555,536 | 4071 | 2 | Acidophile | Soil | [13] |
| P. ananatis PA13 | GCA_000233595.1 | 4,586,378 | 4131 | 1 | Plant pathogen | Rice | [29] |
| P. ananatis LMG 5342 | GCA_000283875.1 | 4,605,545 | 4675 | 1 | N.A. | Human | [30] |
| P. ananatis R100 | GCA_001543055.1 | 4,526,803 | 4353 | 1 | Antagonist against pathogen | Rice | [31] |
| P. ananatis YJ76 | GCA_002224585.2 | 4,671,616 | 4756 | 3 | Growth-promotion | Rice | [32] |
| P. ananatis PNA 97-1R | GCA_002952035.2 | 4,558,720 | 4616 | 2 | Plant pathogen | Onion | [33] |
| P. ananatis NN08200 | GCA_004028255.1 | 4,743,568 | 4598 | 2 | Growth-promotion | Sugarcane | [21] |
| P. ananatis LCFJ-001 | GCA_016598655.1 | 4,499,350 | N.A. | 0 | N.A. | Morus alba | N.A. |
| P. ananatis TZ39 | GCA_019720835.1 | 4,483,976 | 4282 | 2 | Plant pathogen | Rice | [34] |
N.A.: not applicable.
3. Isolation of P. ananatis AJ 13355 and Genome-Editing Tools
In the 1990s, researchers at Ajinomoto Co., Inc. developed L-glutamate fermentation under acidic conditions. The host needed to grow at low pH and resist high L-glutamate concentrations. In the mid-1990s, specialists from Ajinomoto Co., Inc. (Kawasaki, Japan) collected a gram-negative acidophilic bacterium from the soil of a tea plantation in Iwata City (Shizuoka, Japan), which was designated as strain AJ13355. After screening various strains for these properties, P. ananatis strain AJ13355 was selected as the host strain for glutamate fermentation under acidic conditions. This bacterium has proven capable of growing on various sugars and organic acids at acidic and neutral pH values and is resistant to high concentrations of L-glutamic acid [35]. The effects of pH on the specific growth rates of C. glutamicum, E. coli, and P. ananatis are shown in Figure 1.
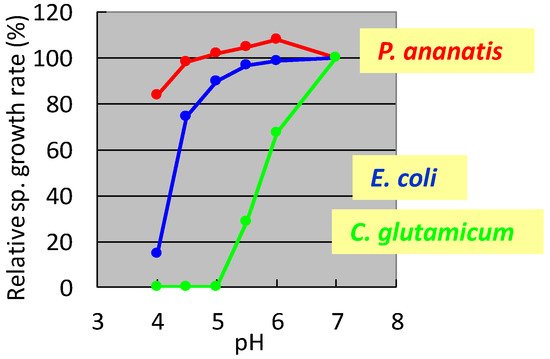
Figure 1. Effect of pH on the specific growth rate of Corynebacterium glutamicum, Escherichia coli, and Pantoea ananatis. The growth rate of each strain at pH 7 was 100%. C. glutamicum: green, E. coli; blue, P. ananatis: red. The data are typical results from three or more independent experiments.
It was shown that C. glutamicum showed good growth only around neutral pH, while P. ananatis showed better growth than E. coli in acidic pH. According to standard microbiological tests on its bacteriological properties and nucleotide sequencing of its 16S rRNA [36], this strain was identified as P. ananatis [37]. The electron micrograph of P. ananatis AJ13355 shows the features of gram-negative and rod-shaped bacteria (Figure 2).
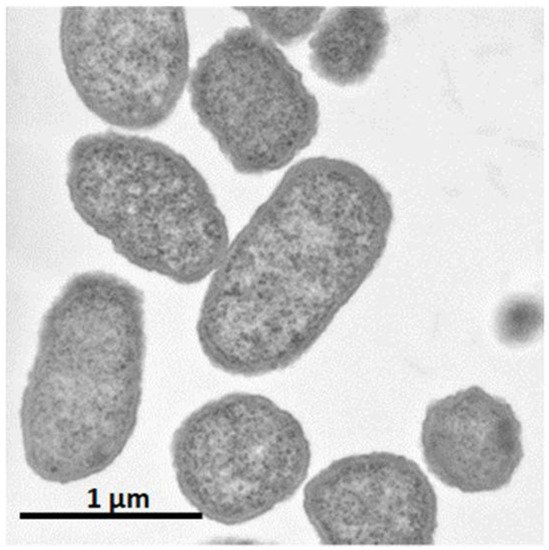
Figure 2. Electron micrograph of Pantoea ananatis AJ13355 at a magnification of ×30,200.
The P. ananatis AJ13355 strain has passed all tests required for industrialization and has been recognized as a biosafety level 1 strain. The complete genomic sequence of P. ananatis AJ13355 was determined and found to consist of a single circular chromosome consisting of 4,555,536 bp (DDBJ: AP012032) and a circular plasmid (pEA320) with 321,744 bp (DDBJ: AP012033). After automated annotation, 4071 protein-coding sequences were identified in the P. ananatis AJ13355 genome [13]. The P. ananatis AJ13355 genome was compared with that of Escherichia coli, a model organism belonging to the Enterobacteriaceae family to which P. ananatis once belonged and which was utilized in the industrial production of various substances. Short colinear regions, which are identical to the DNA sequences in the E. coli MG1655 chromosome, were widely dispersed along the P. ananatis AJ13355 genome. Conjugal gene transfer from E. coli to P. ananatis, mediated by homologous recombination between short identical sequences, has also been experimentally demonstrated [13].
To develop a general genetic tool that can be used in P. ananatis AJ13355, Katashkina et al. [38] constructed novel non-mobilizable derivatives of RSF1010, a well-studied broad-host-range plasmid lacking all known DNA sequences involved in the mobilization process because of the exploitation of λ Red-driven recombination between the plasmid and an in vitro-constructed linear DNA fragment. Mobilization of the obtained RSFmob plasmid was not detected in standard tests, and high stability was demonstrated in E. coli and P. ananatis, which satisfies the biosafety requirements of genetically modified organisms used in scaled-up production [38].
To develop highly active producer strains with metabolically engineered pathways, it is necessary to manipulate many genes and express them individually at different levels or under separate regulatory controls. The construction of plasmid-less marker-less strains using sequential chromosome modifications, including deletions and integration of genetic material, has many advantages for further practical exploitation of these bacteria in industry. The λ Red-recombineering technique previously developed in E. coli is a high-performance tool for the rapid construction of precise genome modifications, such as deletion or insertion of genetic material, nucleotide changes, modification of regulatory regions, and construction of unmarked mutations. Although the expression of λ Red genes in P. ananatis was highly toxic, a mutant strain, SC17(0), grew well under simultaneous expression of λ gam, bet, and exo genes. Using this strain, site-specific and homologous recombination of phage λ, together with the possible transfer of marked mutations by electroporation of the chromosomal DNA, were adapted to genetically engineer P. ananatis AJ13355 and its derivatives [39]. Minaeva et al. [40] developed a new method of foreign DNA insertion for the step-by-step construction of plasmid-less marker-less recombinant E. coli strains with a chromosome structure designed in advance. The dual-in/out strategy is based on the initial Red-driven insertion of artificial φ80-attB sites into the desired points of the chromosome followed by two site-specific recombination processes: first, the φ80 system is used for integration of the recombinant DNA based on selective marker-carrier conditionally replicated plasmid with φ80-attP-site; and, second, the λ system is used for excision of the inserted vector part, including the plasmid ori-replication and the marker, flanked by λ-attL/R-sites [40]. Andreeva et al. [41] adopted a dual-in/out recombineering-driven strategy to integrate DNA fragments into targeted points on the E. coli chromosome for application in P. ananatis. P. ananatis pqqABCDEF was cloned in vivo and integrated into the chromosomes of P. ananatis using the dual-in/out strategy. The introduction of a second copy of pqqABCDEF to P. ananatis SC17(0) doubled the accumulation of PQQ [41]. This new approach has facilitated the design of recombinant marker-less and plasmid-less strains. It allows for the inclusion of large artificial inserts that are difficult to introduce using commonly used PCR-based recombination procedures. Katashkina et al. [42] further improved the dual-in/out system to a method that simultaneously incorporates a few DNA fragments into specific loci on the genome. They divided the mevalonic acid pathway into acetyl-CoA to mevalonic acid (MVA), MVA to phosphomevalonate, and the remaining pathway (see Isoprenoid production). Through the improved dual-in/out system and electroporation efficiency in P. ananatis, they were able to handle plasmids for all three parts simultaneously. They showed that their experimental design was sufficient to remove all selection markers [42]. Consequently, it is now possible to evaluate the production of genetically engineered strains that retain the desired multiple genetic traits, resulting in an increase in breeding speed.
4. L-Cysteine Production
L-Cysteine is an important amino acid with many applications in various industries, including pharmaceuticals, food, and cosmetics. Bacterial fermentation is well-established for many major L-amino acids in commercial production; however, L-cysteine is one of the few exceptions, for which fermentative production methods have been demonstrated only in recent years and are still under development [43][44]. Because of the cytotoxicity of L-cysteine, bacterial cells are equipped with several modes of stringent metabolic regulation that allow them to strictly control the intracellular levels of L-cysteine, making overproduction of this compound more challenging. Dissecting these multifaceted and complicated intracellular regulations and freeing them from negative feedback controls is essential to achieve efficient overproduction. These regulatory mechanisms include feedback inhibition of two key enzymes, serine acetyltransferase (SAT) and 3-phosphoglycerate dehydrogenase (3-PGDH) by L-cysteine and L-serine, respectively, along with the L-cysteine biosynthetic pathway. CysB, a master regulator of sulfur assimilation and L-cysteine metabolism, controls the expression of most of the biosynthetic and metabolic genes associated with L-cysteine at the transcriptional level (Figure 3).
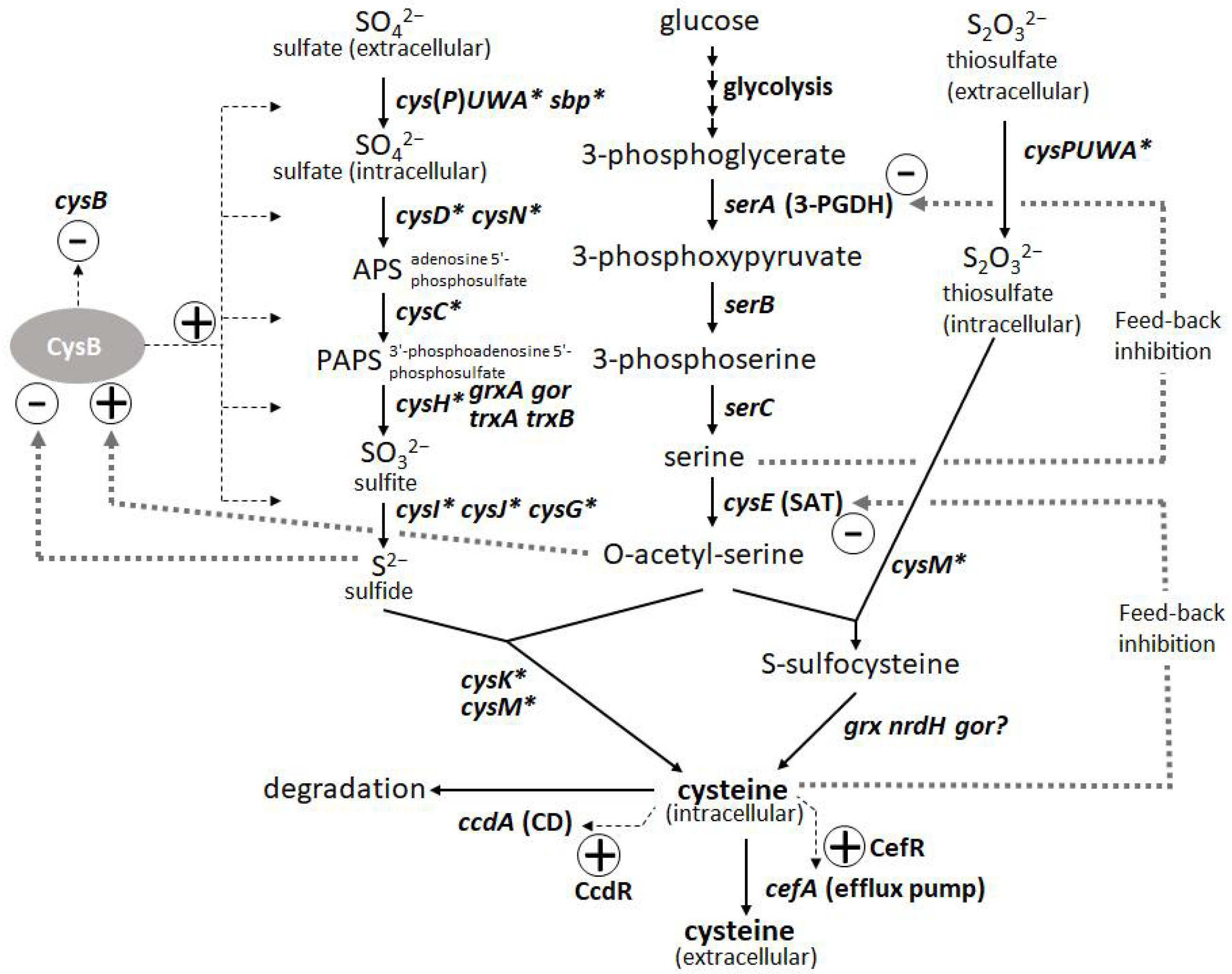
Figure 3. Biosynthetic pathway and regulation of L-cysteine in Pantoea ananatis AJ13355 [45]. Thin and thick dotted lines indicate transcription and protein regulation, respectively. Asterisk indicates CysB regulon.
The degradation of L-cysteine provides another mode of regulation wherein cysteine desulfhydrases (CDs) play a central role in protecting bacterial cells from intracellular over-accumulation. The efflux systems of L-cysteine by specific exporters add an additional layer of regulation, in which they work as a safety valve to cope with the rapid increase in its intracellular levels. Thus, L-cysteine levels are regulated by its biosynthesis, degradation, and efflux [45]. To achieve overproduction, negative regulations, namely SAT and 3-PGDH, must be deregulated, and positive regulations, namely CysB regulons of biosynthesis, must be enhanced. CDs have to be removed, but they must be combined with the enhancement of efflux transporters because the transporters contribute to recovering damaged cells by accumulating L-cysteine under deficient conditions of CDs and promoting extracellular production of L-cysteine as its oxidized form L-cystine (L-cysteine can be easily oxidized extracellularly during aerobic cultivation). Once these core factors specific to L-cysteine production are regulated, general approaches such as enhancing bottlenecks and their biosynthetic pathways and shutting down the pathways to byproducts become more effective [15]. To date, among many amino-acid-producing microbes, P. ananatis and E. coli are advanced bacterial hosts for L-cysteine production [15][46].
The introduction of mutated key biosynthetic enzymes that are free from feedback inhibition is the first step in the development of amino-acid-producing microbes. There are two key enzymes in the L-cysteine biosynthetic pathway in P. ananatis: SAT and 3-PGDH encoded by cysE and serA, respectively. Mutations that remove the feedback inhibition by L-cysteine have been well-studied and applied to L-cysteine production in E. coli (Figure 3). Effective mutations identified for 3-PGDH in E. coli [47] could apply to the corresponding position of this enzyme in P. ananatis [48]. Kai et al. [49] identified many substantial effective mutations in the SAT of E. coli and P. ananatis that abolished feedback inhibition by comparing the crystal structures of SAT with and without the allosteric inhibitor. The basic preliminary L-cysteine-producing strains can be constructed using these mutated enzymes, which typically produce traces of L-cysteine in productive media.
The second major step in genetic engineering is identifying and removing the genes involved in L-cysteine degradation. In E. coli, at least five CDs (TnaA, MetC, MalY, CysK, and CysM [50][51]) and one cysteine desulfidase (YhaM [52][53]) have been identified and demonstrated to exert positive effects on L-cysteine production through gene deletion. While E. coli has developed rather complicated degradation mechanisms involving multiple degradation enzymes, P. ananatis possesses the only CD encoded by ccdA. CcdA in P. ananatis is the major and strongest CD that is induced intensively by L-cysteine using CcdR encoded adjacent to ccdA in the corresponding locus; therefore, this CD is proposed to have a more distinctive function in L-cysteine decomposition to cope with the sudden increase in both intracellular and extracellular L-cysteine levels [54]. Deleting ccdA in P. ananatis is very effective for producing L-cysteine, as it removes detectable levels of CD activity in cells [15][45]. P. ananatis may have advantages over E. coli in managing degradation activity. E. coli strains with multiple gene knockouts could still exhibit significant CD activity [51], indicating that there were additional unidentified CDs that presumably negatively affected L-cysteine production. Moreover, many of the CDs in E. coli have significant metabolic functions (e.g., TnaA is a tryptophanase, and CysM and CysK are cysteine synthases), whose deletion may affect essential physiological functions, including growth and biosynthesis of L-cysteine.
The third step was to identify and utilize the efflux system of L-cysteine. Because of the deletion of CD gene(s), the cells exhibited poor growth that was due to the increased intracellular levels of L-cysteine. The introduction of efflux pumps increases the production of L-cysteine and recovers the damaged cells from poor growth caused by accumulated L-cysteine. YdeD and YfiK are functional efflux pumps in E. coli for L-cysteine [54][55]. However, since they were identified while searching for factors effective for overproducing L-cysteine, their substrates and physiological function in L-cysteine metabolism and regulation are unclear. However, P. ananatis has developed more specific efflux pumps of L-cysteine, associated with its metabolism and regulation. CefA and CefB of P. ananatis were discovered by screening for factors that confer resistance to high concentrations of L-cysteine (Figure 3). CefA was found to be inducible by L-cysteine using its counterpart regulator CefR, encoded adjacent to cefA in the corresponding locus; therefore, it has been proposed that it functions as a specific safety valve of L-cysteine [45]. Both CefA and CefB were demonstrated to be involved in L-cysteine overproduction in P. ananatis, where they significantly contributed to high-level production and recovery from L-cysteine toxicity caused by the absence of its decomposer ccdA [15]. P. ananatis may have an advantage over E. coli in exporting L-cysteine because it possesses more specific and efficient efflux systems that may allow the export of L-cysteine specifically without leaking out some essential metabolites required for cell viability.
References
- Coutinho, T.A.; Venter, S.N. Pantoea ananatis: An Unconventional Plant Pathogen. Mol. Plant Pathol. 2009, 10, 325–335.
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, R.S. Genome-Based Phylogeny and Taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. Divided into the Families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599.
- Serrano, F.B. Bacterial Fruitlet Brown-Rot of Pineapple in the Philippines. Philipp. J. Sci. 1928, 36, 271–324.
- Gavini, F.; Mergaert, J.; Beji, A.; Mielcarek, C.; Izard, D.; Kersters, K.; De Ley, J. Transfer of Enterobacter agglomerans (Beijerinck 1888) Ewing and Fife 1972 to Pantoea gen. nov. as Pantoea agglomerans comb. nov. and Description of Pantoea dispersa sp. nov. Int. J. Syst. Bacteriol. 1989, 39, 337–345.
- Mergaert, J.; Verdonck, L.; Kersters, K. Transfer of Erwinia ananas (Synonym, Erwinia uredovora) and Erwinia stewartii to the Genus Pantoea emend. as Pantoea ananas (Serrano 1928) comb. nov. and Pantoea stewartii (Smith 1898) comb. nov., Respectively, and Description of Pantoea stewartii subsp. indologenes subsp. nov. Int. J. Syst. Bacteriol. 1993, 43, 162–173.
- Trüper, H.G.; De’Clari, L. Taxonomic Note: Necessary Correction of Specific Epithets Formed as Substantives (Nouns) “in Apposition”. Int. J. Syst. Evol. 1997, 47, 908–909.
- Weller-Stuart, T.; De Maayer, P.; Coutinho, T. Pantoea ananatis: Genomic Insights into a Versatile Pathogen. Mol. Plant Pathol. 2017, 18, 1191–1198.
- Becker, J.; Wittmann, C. Advanced Biotechnology: Metabolically Engineered Cells for the Bio-Based Production of Chemicals and Fuels, Materials, and Health-Care Products. Angew. Chem. Int. Ed. Engl. 2015, 54, 3328–3350.
- Pontrelli, S.; Chiu, T.Y.; Lan, E.I.; Chen, F.Y.; Chang, P.; Liao, J.C. Escherichia coli as a Host for Metabolic Engineering. Metab. Eng. 2018, 50, 16–46.
- Kinoshita, S.; Udaka, S.; Shimono, M. Studies on The Amino Acid Fermentation. J. Gen. Appl. Microbiol. 1957, 3, 193–205.
- Becker, J.; Wittmann, C. Bio-Based Production of Chemicals, Materials and Fuels—Corynebacterium glutamicum as Versatile Cell Factory. Curr. Opin. Biotechnol. 2012, 23, 631–640.
- Tsuge, Y.; Matsuzawa, H. Recent Progress in Production of Amino Acid-Derived Chemicals Using Corynebacterium glutamicum. World J. Microbiol. Biotechnol. 2021, 37, 49.
- Hara, Y.; Kadotani, N.; Izui, H.; Katashkina, J.I.; Kuvaeva, T.M.; Andreeva, I.G.; Golubeva, L.I.; Malko, D.B.; Makeev, V.J.; Mashko, S.V.; et al. The Complete Genome Sequence of Pantoea ananatis AJ13355, an Organism with Great Biotechnological Potential. Appl. Microbiol. Biotechnol. 2012, 93, 331–341.
- Usuda, Y.; Hara, Y.; Kojima, H. Toward Sustainable Amino Acid Production. In Advances in Biochemical Engineering/Biotechnology; Yokota, A., Ikeda, M., Eds.; Springer: Tokyo, Japan, 2017; Volume 159, pp. 289–304.
- Takumi, K.; Ziyatdinov, M.K.; Samsonov, V.; Nonaka, G. Fermentative Production of Cysteine by Pantoea ananatis. Appl. Environ. Microbiol. 2017, 83, e02502-16.
- Nitta, N.; Tajima, Y.; Katashkina, J.I.; Yamamoto, Y.; Onuki, A.; Rachi, H.; Kazieva, E.; Nishio, Y. Application of Inorganic Phosphate Limitation to Efficient Isoprene Production in Pantoea ananatis. J. Appl. Microbiol. 2020, 128, 763–774.
- Nitta, N.; Tajima, Y.; Yamamoto, Y.; Moriya, M.; Matsudaira, A.; Hoshino, Y.; Nishio, Y.; Usuda, Y. Fermentative Production of Enantiopure (S)-Linalool Using a Metabolically Engineered Pantoea ananatis. Microb. Cell Fact. 2021, 20, 54.
- Thomas, P.; Kumari, S.; Swarna, G.K.; Gowda, T.K.S. Papaya Shoot Tip Associated Endophytic Bacteria Isolated from In Vitro Cultures and Host–Endophyte Interaction In Vitro and In Vivo. Can. J. Microbiol. 2007, 53, 380–390.
- Kim, S.-N.; Cho, W.K.; Kim, W.-I.; Jee, H.J.; Park, C.-S. Growth Promotion of Pepper Plants by Pantoea ananatis B1-9 and Its Efficient Endophytic Colonization Capacity in Plant Tissues. Plant Pathol. J. 2012, 28, 270–281.
- Shi, G.-Y.; Zeng, Q.; Nong, Z.M.; Ye, X.-L.; Cen, Z.-L.; Li, Y.-R.; Hu, C.-J. Identification of an Endophytic Nitrogen-Fixing Bacterium NN08200 from Sugarcane and Its Growth Promotion of Sugarcane. Microbiol. China. 2019, 46, 1336–1345.
- Zeng, Q.; Shi, G.; Nong, Z.; Ye, X.; Hu, C. Complete Genome Sequence of Pantoea ananatis strain NN08200, an Endophytic Bacterium Isolated from Sugarcane. Curr. Microbiol. 2020, 77, 1864–1870.
- Gkorezis, P.; Van Hamme, J.D.; Bottos, E.M.; Thijs, S.; Balseiro-Romero, M.; Monterroso, C.; Kidd, P.S.; Rineau, F.; Weyens, N.; Vangronsveld, J. Draft Genome Sequence of Pantoea ananatis GB1, a Plant-Growth-Promoting Hydrocarbonoclastic Root Endophyte, Isolated at a Diesel Fuel Phytoremediation Site Planted with Populus. Genome Announc. 2016, 4, e00028-16.
- Megías, E.; Megías, M.; Ollero, F.J.; Hungria, M. Draft Genome Sequence of Pantoea ananatis strain AMG521, a Rice Plant Growth-Promoting Bacterial Endophyte Isolated from the Guadalquivir Marshes in Southern Spain. Genome Announc. 2016, 4, e01681-15.
- Megías, E.; Dos Reis Junior, F.B.; Ribeiro, R.A.; Ollero, F.J.; Megías, M.; Hungria, M. Draft Genome Sequence of Pantoea ananatis strain 1.38, a Bacterium Isolated from the Rhizosphere of Oryza sativa var. Puntal That Shows Biotechnological Potential as an Inoculant. Genome Announc. 2018, 6, e01547-17.
- Kim, H.J.; Lee, J.H.; Kang, B.R.; Rong, X.; McSpadden Gardener, B.B.; Ji, H.J.; Park, C.S.; Kim, Y.C. Draft Genome Sequence of Pantoea ananatis B1-9, a Nonpathogenic Plant Growth-Promoting Bacterium. J. Bacteriol. 2012, 194, 729.
- Misawa, N.; Shimada, H. Metabolic Engineering for the Production of Carotenoids in non-Carotenogenic Bacteria and Yeasts. J. Biotechnol. 1997, 59, 169–181.
- Yoon, S.H.; Kim, J.E.; Lee, S.H.; Park, H.M.; Choi, M.S.; Kim, J.Y.; Lee, S.H.; Shin, Y.C.; Keasling, J.D.; Kim, S.W. Engineering the Lycopene Synthetic Pathway in E. coli by Comparison of the Carotenoid Genes of Pantoea agglomerans and Pantoea ananatis. Appl. Microbiol. Biotechnol. 2007, 74, 131–139.
- De Maayer, P.; Chan, W.Y.; Venter, S.N.; Toth, I.K.; Birch, P.R.; Joubert, F.; Coutinho, T.A. Genome Sequence of Pantoea ananatis LMG20103, the Causative Agent of Eucalyptus Blight and Dieback. J. Bacteriol. 2010, 192, 2936–2937.
- Choi, O.; Lim, J.Y.; Seo, Y.S.; Hwang, I.; Kim, J. Complete Genome Sequence of the Rice Pathogen Pantoea ananatis strain PA13. J. Bacteriol. 2012, 194, 531.
- De Maayer, P.; Chan, W.Y.; Rezzonico, F.; Bühlmann, A.; Venter, S.N.; Blom, J.; Goesmann, A.; Frey, J.E.; Smits, T.H.; Duffy, B.; et al. Complete Genome Sequence of Clinical Isolate Pantoea ananatis LMG 5342. J. Bacteriol. 2012, 194, 1615–1616.
- Wu, L.; Liu, R.; Niu, Y.; Lin, H.; Ye, W.; Guo, L.; Hu, X. Whole Genome Sequence of Pantoea ananatis R100, an Antagonistic Bacterium Isolated from Rice Seed. J. Biotechnol. 2016, 225, 1–2.
- Zheng, J.; Yu, J.; Jia, M.; Zheng, L.; Feng, Y. Indole Enhances the Survival of Pantoea ananatis YJ76 in Face of Starvation Conditions. J. Basic Microbiol. 2017, 57, 633–639.
- Stice, S.P.; Stumpf, S.D.; Gitaitis, R.D.; Kvitko, B.H.; Dutta, B. Pantoea ananatis Genetic Diversity Analysis Reveals Limited Genomic Diversity as Well as Accessory Genes Correlated with Onion Pathogenicity. Front. Microbiol. 2018, 9, 184.
- Yu, L.; Yang, C.; Ji, Z.; Zeng, Y.; Liang, Y.; Hou, Y. First Report of New Bacterial Leaf Blight of Rice Caused by Pantoea ananatis in Southeast China. Plant Dis. 2021, 106, PDIS-05.
- Moriya, M.; Izui, H.; Ono, E.; Matsui, K.; Ito, H.; Hara, Y. L-Glutamic Acid-Producing Bacterium and Method for Producing L-Glutamic Acid. U.S. Patent 6,331,419, 18 December 2001.
- Kwon, S.W.; Go, S.J.; Kang, H.W.; Ryu, J.C.; Jo, J.K. Phylogenetic Analysis of Erwinia species Based on 16S RRNA Gene Sequences. Int. J. Syst. Bacteriol. 1997, 47, 1061–1067.
- Takahashi, Y.; Tateyama, Y.; Sato, M. Process for Producing L-Glutamic Acid. U.S. Patent US7,354,744, 8 April 2008.
- Katashkina, J.I.; Kuvaeva, T.M.; Andreeva, I.G.; Skorokhodova, A.Y.; Biryukova, I.V.; Tokmakova, I.L.; Golubeva, L.I.; Mashko, S.V. Construction of Stably Maintained Non-Mobilizable Derivatives of RSF1010 Lacking All Known Elements Essential for Mobilization. BMC Biotechnol. 2007, 7, 80.
- Katashkina, J.I.; Hara, Y.; Golubeva, L.I.; Andreeva, I.G.; Kuvaeva, T.M.; Mashko, S.V. Use of the λ Red-Recombineering Method for Genetic Engineering of Pantoea ananatis. BMC Mol. Biol. 2009, 10, 34.
- Minaeva, N.I.; Gak, E.R.; Zimenkov, D.V.; Skorokhodova, A.Y.; Biryukova, I.V.; Mashko, S.V. Dual-In/Out Strategy for Genes Integration into Bacterial Chromosome: A Novel Approach to Step-by-Step Construction of Plasmid-Less Marker-Less Recombinant E. coli Strains with Predesigned Genome Structure. BMC Biotechnol. 2008, 8, 63.
- Andreeva, I.G.; Golubeva, L.I.; Kuvaeva, T.M.; Gak, E.R.; Katashkina, J.I.; Mashko, S.V. Identification of Pantoea ananatis Gene Encoding Membrane Pyrroloquinoline Quinone (PQQ)-Dependent Glucose Dehydrogenase and pqqABCDEF Operon Essential for PQQ Biosynthesis. FEMS Microbiol. Lett. 2011, 318, 55–60.
- Katashkina, J.I.; Kazieva, E.D.; Tajima, Y.; Mashko, S.V. Increased Isoprene Production by the Recombinant Pantoea ananatis strain due to the Balanced Amplification of Mevalonate Pathway Genes. Appl. Biochem. Microbiol. 2019, 55, 850–860.
- Wada, M.; Takagi, H. Metabolic Pathways and Biotechnological Production of L-Cysteine. Appl. Microbiol. Biotechnol. 2006, 73, 48–54.
- Takagi, H.; Ohtsu, I. L-Cysteine Metabolism and Fermentation in Microorganisms. Adv. Biochem. Eng. Biotechnol. 2017, 159, 129–151.
- Takumi, K.; Nonaka, G. Bacterial Cysteine-Inducible Cysteine Resistance Systems. J. Bacteriol. 2016, 198, 1384–1392.
- Liu, H.; Hou, Y.; Wang, Y.; Li, Z. Enhancement of Sulfur Conversion Rate in the Production of L-Cysteine by Engineered Escherichia coli. J. Agric. Food Chem. 2020, 68, 250–257.
- Al-Rabiee, R.; Zhang, Y.; Grant, G.A. The Mechanism of Velocity Modulated Allosteric Regulation in D-3-phosphoglycerate Dehydrogenase. Site-Directed Mutagenesis of Effector Binding Site Residues. J. Biol. Chem. 1996, 271, 23235–23238.
- Takumi, K.; Nonaka, G. An L-Amino Acid-Producing Bacterium and a Method for Producing an L-Amino Acid. European Patent EP2,218,729, 9 July 2014.
- Kai, Y.; Kashiwagi, T.; Ishikawa, K.; Ziyatdinov, M.K.; Redkina, E.I.; Kiriukhin, M.Y.; Gusyatiner, M.M.; Kobayashi, S.; Takagi, H.; Suzuki, E.E. Engineering of Escherichia coli L-Serine O-acetyltransferase on the Basis of Crystal Structure: Desensitization to Feedback Inhibition by L-Cysteine. Protein Eng. Des. Sel. 2006, 19, 163–167.
- Awano, N.; Wada, M.; Kohdoh, A.; Oikawa, T.; Takagi, H.; Nakamori, S. Effect of Cysteine Desulfhydrase Gene Disruption on L-Cysteine Overproduction in Escherichia coli. Appl. Microbiol. Biotechnol. 2003, 62, 239–243.
- Awano, N.; Wada, M.; Mori, H.; Nakamori, S.; Takagi, H. Identification and Functional Analysis of Escherichia coli Cysteine Desulfhydrases. Appl. Environ. Microbiol. 2005, 71, 4149–4152.
- Shimada, T.; Tanaka, K.; Ishihama, A. Transcription Factor DecR (YbaO) Controls Detoxification of L-Cysteine in Escherichia coli. Microbiology 2016, 162, 1698–1707.
- Nonaka, G.; Takumi, K. Cysteine Degradation Gene yhaM, Encoding Cysteine Desulfidase, Serves as a Genetic Engineering Target to Improve Cysteine Production in Escherichia coli. AMB Express 2017, 7, 90.
- Dassler, T.; Maier, T.; Winterhalter, C.; Böck, A. Identification of a Major Facilitator Protein from Escherichia coli Involved in Efflux of Metabolites of the Cysteine Pathway. Mol. Microbiol. 2000, 36, 1101–1112.
- Franke, I.; Resch, A.; Dassler, T.; Maier, T.; Böck, A. YfiK from Escherichia coli Promotes Export of O-Acetylserine and Cysteine. J. Bacteriol. 2003, 185, 1161–1166.
More
Information
Subjects:
Biotechnology & Applied Microbiology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
23 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No