Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yoshihiko Hara and Version 2 by Conner Chen.
Pantoea ananatis, a gram-negative bacterium belonging to the Erwiniaceae family, is a well-known phytopathogen isolated from many ecological niches and plant hosts. However, this bacterium also provides us with various beneficial characteristics, such as the growth promotion of their host plants and increased crop yield.
- Pantoea ananatis
- microbial production
1. Introduction
Pantoea ananatis is a gram-negative, rod-shaped, aerobic, or facultatively anaerobic bacteria belonging to the class Gammaproteobacteria and was recently reclassified into the family Erwiniaceae from Enterobacteriaceae [1][2][1,2]. P. ananatis was first described as Erwinia ananas by Serrano [3]. Some strains of Enterobacter agglomerans, Erwinia herbicola, and Erwinia milletiae that form part of the E. herbicola–E. agglomerans complex was assigned to the genus Pantoea [4]. Later, Pantoea uredovora, a pathogen of Puccinia graminis, was shown to have a high level of genomic relatedness to P. ananas, and the two species were synonymized [5]. P. ananas proposed by Mergaert et al. [5] was corrected to P. ananatis by Trüper and De’Clari [6]. Due to this phylogenetic history, P. ananatis contains various kinds of plant pathogenic strains [1]. At the same time, strains that promote plant growth that are applicable to bioremediation, or have lignocellulose degradation capacity, have been found in recent years [7].
In the field of microbial biomanufacturing, Escherichia coli, a model organism and a member of the family Enterobacteriaceae, which is a gram-negative facultative anaerobic rod, has been used industrially for the production of various substances because of its high growth rate and sugar consumption activity in the neutral pH range [8][9][8,9]. Corynebacterium glutamicum, a gram-positive, rod-shaped bacterium, is capable of L-glutamate fermentation [10] and has been used for the industrial production of many substances by taking advantage of its characteristic cell surface [11][12][11,12]. To date, bacterial fermentation production has been realized by mainly using E. coli and C. glutamicum, and many tools for genetic engineering have been developed for both strains [9]. However, it was clear that if both strains had the trait of robustness to pH, they would be more desirable industrial substance-producing bacteria. P. ananatis AJ13355 was isolated as an acidophilic bacterium [13] and has been used as an excellent host organism in previous studies to produce amino acids such as L-glutamate [14], L-cysteine [15], and isoprenoids [16][17][16,17]. The emergence of strains closely related to E. coli and advantageous for industrial production is important in terms of increasing the potential for substance production.
2. Characteristic of Pantoea ananatis
P. ananatis has been isolated from various environments and hosts and is well-known for its phytopathogenicity. P. ananatis causes disease symptoms in many economically important agronomic crops and forest tree species worldwide [1]. However, several strains have been known to improve the growth of plants [7], such as papaya [18], red pepper [19], sugarcane [20][21][20,21], poplar [22], and rice [23][24][23,24]. Kim et al. [19][25][19,25] determined the genome sequence of P. ananatis B1–9 isolated from the rhizosphere of the green onion and reported that enhanced red pepper crop yield by approximately three times and showed phosphate solubilization, sulfur oxidation, nitrogen fixation, and indole-3-acetic acid (IAA) production activities. P. ananatis AMG521, isolated as an endophyte from rice paddies, showed the capacity to synthesize siderophores, cellulose, and IAA, and the capacity to solubilize phosphate and increase rice yield [23]. P. ananatis strain 1.38, isolated from the rhizosphere of rice (Oryza sativa L.), has been reported to have phosphate solubilization activity, siderophore and auxin production, and cellulose, lipase, and pectinase activities [24]. It has long been known that P. ananatis produces carotenoids, and it has been reported that introducing the carotenoid biosynthesis genes of P. ananatis into a microorganism that does not produce carotenoids leads to the production of lycopene, astaxanthin, and β-carotene [26][27][26,27]. Ten complete genomes of P. ananatis have been determined and registered. Four were pathogenic and four had useful traits (Table 1).Table 1.
Complete genome of
Pantoea ananatis
.
| Strain | Assembly | Chromosome Size | CDS | Plasmids | Classification | Source | Reference |
|---|---|---|---|---|---|---|---|
| Pantoea ananatis LMG 20103 | GCA_000025405.2 | 4,703,373 | 4272 | 0 | Plant pathogen | Eucalyptus | [28] |
| P. ananatis AJ13355 | GCA_000270125.2 | 4,555,536 | 4071 | 2 | Acidophile | Soil | [13] |
| P. ananatis PA13 | GCA_000233595.1 | 4,586,378 | 4131 | 1 | Plant pathogen | Rice | [29] |
| P. ananatis LMG 5342 | GCA_000283875.1 | 4,605,545 | 4675 | 1 | N.A. | Human | [30] |
| P. ananatis R100 | GCA_001543055.1 | 4,526,803 | 4353 | 1 | Antagonist against pathogen | Rice | [31] |
| P. ananatis YJ76 | GCA_002224585.2 | 4,671,616 | 4756 | 3 | Growth-promotion | Rice | [32] |
| P. ananatis PNA 97-1R | GCA_002952035.2 | 4,558,720 | 4616 | 2 | Plant pathogen | Onion | [33] |
| P. ananatis NN08200 | GCA_004028255.1 | 4,743,568 | 4598 | 2 | Growth-promotion | Sugarcane | [21] |
| P. ananatis LCFJ-001 | GCA_016598655.1 | 4,499,350 | N.A. | 0 | N.A. | Morus alba | N.A. |
| P. ananatis TZ39 | GCA_019720835.1 | 4,483,976 | 4282 | 2 | Plant pathogen | Rice | [34] |
N.A.: not applicable.
3. Isolation of P. ananatis AJ 13355 and Genome-Editing Tools
In the 1990s, researchers at Ajinomoto Co., Inc. developed L-glutamate fermentation under acidic conditions. The host needed to grow at low pH and resist high L-glutamate concentrations. In the mid-1990s, specialists from Ajinomoto Co., Inc. (Kawasaki, Japan) collected a gram-negative acidophilic bacterium from the soil of a tea plantation in Iwata City (Shizuoka, Japan), which was designated as strain AJ13355. After screening various strains for these properties, P. ananatis strain AJ13355 was selected as the host strain for glutamate fermentation under acidic conditions. This bacterium has proven capable of growing on various sugars and organic acids at acidic and neutral pH values and is resistant to high concentrations of L-glutamic acid [35]. The effects of pH on the specific growth rates of C. glutamicum, E. coli, and P. ananatis are shown in Figure 1.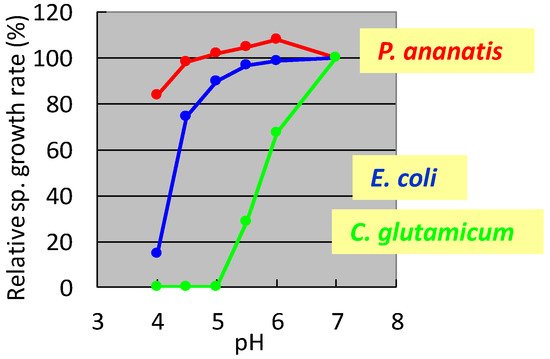
Figure 1. Effect of pH on the specific growth rate of Corynebacterium glutamicum, Escherichia coli, and Pantoea ananatis. The growth rate of each strain at pH 7 was 100%. C. glutamicum: green, E. coli; blue, P. ananatis: red. The data are typical results from three or more independent experiments.
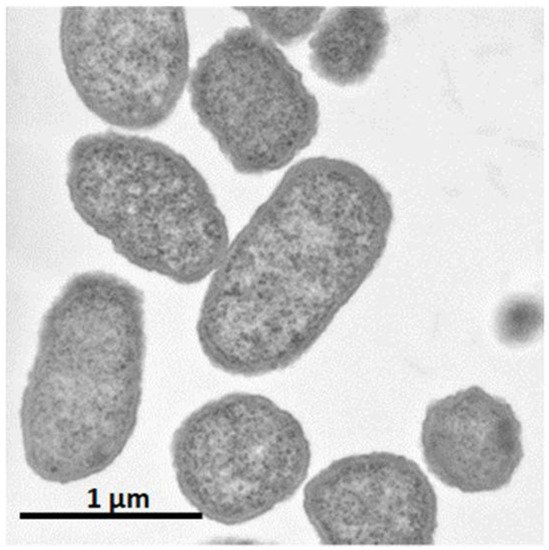
Figure 2.
Electron micrograph of
Pantoea ananatis
AJ13355 at a magnification of ×30,200.
4. L-Cysteine Production
L-Cysteine is an important amino acid with many applications in various industries, including pharmaceuticals, food, and cosmetics. Bacterial fermentation is well-established for many major L-amino acids in commercial production; however, L-cysteine is one of the few exceptions, for which fermentative production methods have been demonstrated only in recent years and are still under development [43][44][43,44]. Because of the cytotoxicity of L-cysteine, bacterial cells are equipped with several modes of stringent metabolic regulation that allow them to strictly control the intracellular levels of L-cysteine, making overproduction of this compound more challenging. Dissecting these multifaceted and complicated intracellular regulations and freeing them from negative feedback controls is essential to achieve efficient overproduction. These regulatory mechanisms include feedback inhibition of two key enzymes, serine acetyltransferase (SAT) and 3-phosphoglycerate dehydrogenase (3-PGDH) by L-cysteine and L-serine, respectively, along with the L-cysteine biosynthetic pathway. CysB, a master regulator of sulfur assimilation and L-cysteine metabolism, controls the expression of most of the biosynthetic and metabolic genes associated with L-cysteine at the transcriptional level (Figure 3).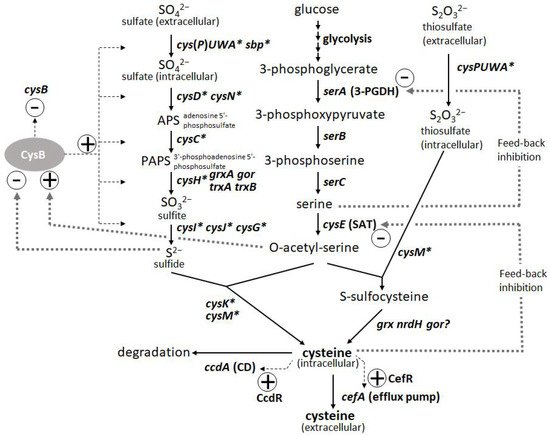
Figure 3. Biosynthetic pathway and regulation of L-cysteine in Pantoea ananatis AJ13355 [45]. Thin and thick dotted lines indicate transcription and protein regulation, respectively. Asterisk indicates CysB regulon.