 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mario Gonzalez-Gronow | -- | 2326 | 2022-06-15 18:24:46 | | | |
| 2 | Peter Tang | Meta information modification | 2326 | 2022-06-16 04:52:01 | | |
Video Upload Options
The 78 kDa glucose-regulated protein (GRP78), a member of the 70 kDa heat-shock family of molecular chaperones (HSP70), is essential for the regulation of the unfolded protein response (UPR) resulting from cellular endoplasmic reticulum (ER) stress. During ER stress, GRP78 evades retention mechanisms and is translocated to the cell surface (csGRP78) where it functions as an autoantigen. Autoantibodies to GRP78 appear in prostate, ovarian, gastric, malignant melanoma, and colorectal cancers. They are also found in autoimmune pathologies such as rheumatoid arthritis (RA), neuromyelitis optica (NMO), anti-myelin oligodendrocyte glycoprotein antibody-associated disorder (AMOGAD), Lambert-Eaton myasthenic syndrome (LEMS), multiple sclerosis (MS), neuropsychiatric systemic lupus erythematosus (NPSLE) and type 1 diabetes (T1D).
1. Introduction
2. Molecular Mechanisms and ER Stress Conditions That Promote GRP78 Autoimmunity in Cancer
3. Mechanisms and ER Stress Conditions That Promote GRP78 Autoimmunity in RA
4. GRP78 Autoantibodies in Immune-Mediated Neurological Diseases
References
- Kiang, J.G.; Tsokos, G.C. Heat shock protein 70 kDa: Molecular biology, biochemistry and physiology. Pharmacol. Ther. 1998, 80, 183–201.
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510.
- Wiersma, V.R.; Michalak, M.; Abdullah, T.M.; Bremer, E.; Eggleton, P. Mechanisms of translocation of ER chaperones to the cell surface and immunomodulatory roles in cancer and autoimmunity. Front. Oncol. 2015, 5, 7.
- Gonzalez-Gronow, M.; Selim, M.A.; Papalas, J.; Pizzo, S.V. GRP78: A multifunctional receptor on the cell surface. Antioxid. Redox Signal. 2009, 11, 2299–2306.
- Raiter, A.; Vilkin, A.; Gingold, R.; Levi, Z.; Halpern, M.; Niv, Y.; Britta-Hardy, B. The presence of anti-GRP78 antibodies in the serum of patients with colorectal carcinoma: A potential biomarker for early cancer detection. Int. J. Biol. Mark. 2014, 43, 1283–1287.
- Bodman-Smith, M.D.; Corrigal, V.M.; Berglin, E.; Cornell, H.R.; Tzioufas, A.G.; Magravani, C.P.; Chan, C.; Rantapää-Dahiqvist, S.; Panayi, G.S. Antibody response to the human stress protein BiP in rheumatoid arthritis. Rheumatology 2004, 43, 1283–1287.
- Shimizu, F.; Takeshita, Y.; Hamamoto, Y.; Yasuteru, Y.; Honda, M.; Sato, R.; Maeda, T.; Takahashi, T.; Fujikawa, S. GRP78 antibodies are associated with clinical phenotype in neuromyelitis optica. Ann. Clin. Transl. Neurol. 2019, 142, 2253–2264.
- Shimizu, F.; Takeshita, Y.; Sano, Y.; Hamamoto, Y.; Shiraishi, H.; Sato, T.; Yoshimura, S.; Maeda, T.; Fujikawa, S.; Nishihara, H.; et al. GRP78 antibodies damage the blood-brain barrier and relate to cerebellar degeneration in Lamber-Eaton myasthenic syndrome. Brain 2019, 142, 2253–2264.
- Rondas, D.; Crèvecoeur, I.; D’Hertrog, W.; Ferreira, G.B.; Staes, A.; Garg, A.D.; Eizirik, D.L.; Agostinis, P.; Gevaert, K.; Overbergh, L.; et al. Citrullinated glucose-regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes 2015, 64, 573–586.
- Shimizu, F.; Nishihara, H.; Kanda, T. Blood-brain barrier dysfunction in immune-mediated neurological diseases. Immunol. Med. 2018, 41, 120–128.
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664.
- Hasnain, S.Z.; Lourie, R.; Das, I.; Chen, A.C.; McGuckin, M.A. The interplay between endoplasmic reticulum stress and inflammation. Immunol. Cell. Biol. 2012, 90, 260–270.
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and immunosuppresive effects of endoplasmic reticulum stress in cancer. Cell 2017, 168, 692–706.
- Park, Y.-J.; Yoo, S.-A.; Kim, W.-U. Role of the endoplasmic reticulum stress in rheumatoid arthritis pathogenesis. J. Korean Med. Sci. 2014, 29, 2–11.
- Gidalevitz, T.; Stevens, F.; Argon, Y. Orchestration of secretory protein folding by ER chaperones. Biochim. Biophys. Acta 2013, 1833, 2410–2424.
- Blond-Elguindi, S.; Fourie, A.M.; Sambrook, J.E.; Gething, M.J. Peptide-dependent stimulation of the ATPase activity of the molecular chaperone BiP is the result of conversion of oligomers to active monomers. J. Biol. Chem. 1993, 268, 12735–12739.
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic resticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169.
- Baumeister, P.; Luo, S.; Skarnes, W.C.; Sui, G.; Seto, E.; Shi, Y.; Lee, A.S. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: Activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 2005, 25, 4529–4540.
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276.
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075.
- Gutiérrez, T.; Simmen, T. Endoplasmic reticulum chaperones and oxidoreductases: Critical regulators of tumor cell survival and immunorecognition. Front. Oncol. 2014, 4, 27–37.
- Gonzalez-Gronow, M.; Cuchacovich, M.; Llanos, C.; Urzua, C.; Gawdi, G.; Pizzo, S.V. Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum. Cancer Res. 2006, 66, 11424–11431.
- Selim, M.A.; Burchette, J.M.; Bowers, E.V.; de Ridder, G.G.; Mo, L.; Pizzo, S.V.; Gonzalez-Gronow, M. Changes in oligosaccharide chains of autoantibodies to GRP78 expressed during progression of malignant melanoma stimulate melanoma cell growth and survival. Melanoma Res. 2011, 21, 323–334.
- Zhang, Y.; Tseng, C.C.; Tsai, Y.L.; Fu, X.; Schiff, R.; Lee, A.S. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI (3, 4, 5) P3 production. PLoS ONE 2013, 8, e80071.
- Liu, R.; Li, X.; Gao, W.; Zhou, Y.; Wey, S.; Mitra, S.K.; Krasnoperov, V.; Dong, D.; Liu, S.; Li, D.; et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin. Cancer Res. 2013, 19, 6802–6811.
- Triantafilou, M.; Fradelizi, D.; Triantafilou, K. Major histocompatibility class one molecule associates with glucose related protein (GRP)78 on the cell surface. Hum. Immunol. 2001, 62, 764–770.
- Ulianich, L.; Terrazzano, G.; Annunziatella, M.; Ruggiero, G.; Beguinot, F.; Di Jeso, B. ER stress impairs MHC class I surface expression and increases susceptibilioty of thyroid cells to NK-mediated cytotoxicity. Biochim. Biophys. Acta 2011, 4, 431–438.
- Marincola, F.M.; Jafee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 2000, 74, 181–273.
- Seliger, B.; Atkins, D.; Bock, M.; Ritz, U.; Ferrone, S.; Huber, C.; Storkel, S. Characterization of human lymphocyte antigen class I antigen-processing machinery defects in renal cell carcinoma lesions with special emphasis on transporter-associated with antigen-processing down-regulation. Clin. Cancer Res. 2003, 9, 1721–1727.
- Mintz, P.J.; Kim, J.; Do, K.A.; Wang, X.; Zinnere, R.G.; Cristofanilli, M.; Arap, M.A.; Hong, W.K.; Logothetis, C.J.; Pasqualini, R.; et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotechnol. 2003, 21, 57–63.
- Misra, U.K.; Gonzalez-Gronow, M.; Gawdi, G.; Wang, F.; Pizzo, S.V. A novel receptor function for the heat shock protein GRP78 gene expression attenuates alpha-2M* induced signaling. Cell Signal. 2004, 16, 929–938.
- Gerçel-Taylor, C.; Bazzett, L.B.; Taylor, D.D. Presence of aberrant tumor-reactive immunoglobulins in the circulation of patients with ovarian cancer. Gynecol. Oncol. 2001, 81, 71–76.
- Kanoh, Y.; Ohara, T.; Mashiko, T.; Abe, T.; Masuda, N.; Akahoski, T. Relationship between N-linked oligosaccharide chains of human serum immunoglobulin G and serum tumor markers with non-small cell lung cancer progression. Anticancer Res. 2006, 26, 4293–4298.
- Kanoh, Y.; Ohara, T.; Tadano, T.; Kanoh, M.; Akahoski, T. Changes to N-linked oligosaccharide chains of human serum immunoglobulin G and matrix metalloproteinase-2 with cancer progression. Anticancer Res. 2008, 28, 715–720.
- Margni, R.A.; Borel, I.M. Paradoxical behavior of asymmetric IgG antibodies. Immunol. Rev. 1998, 163, 77–87.
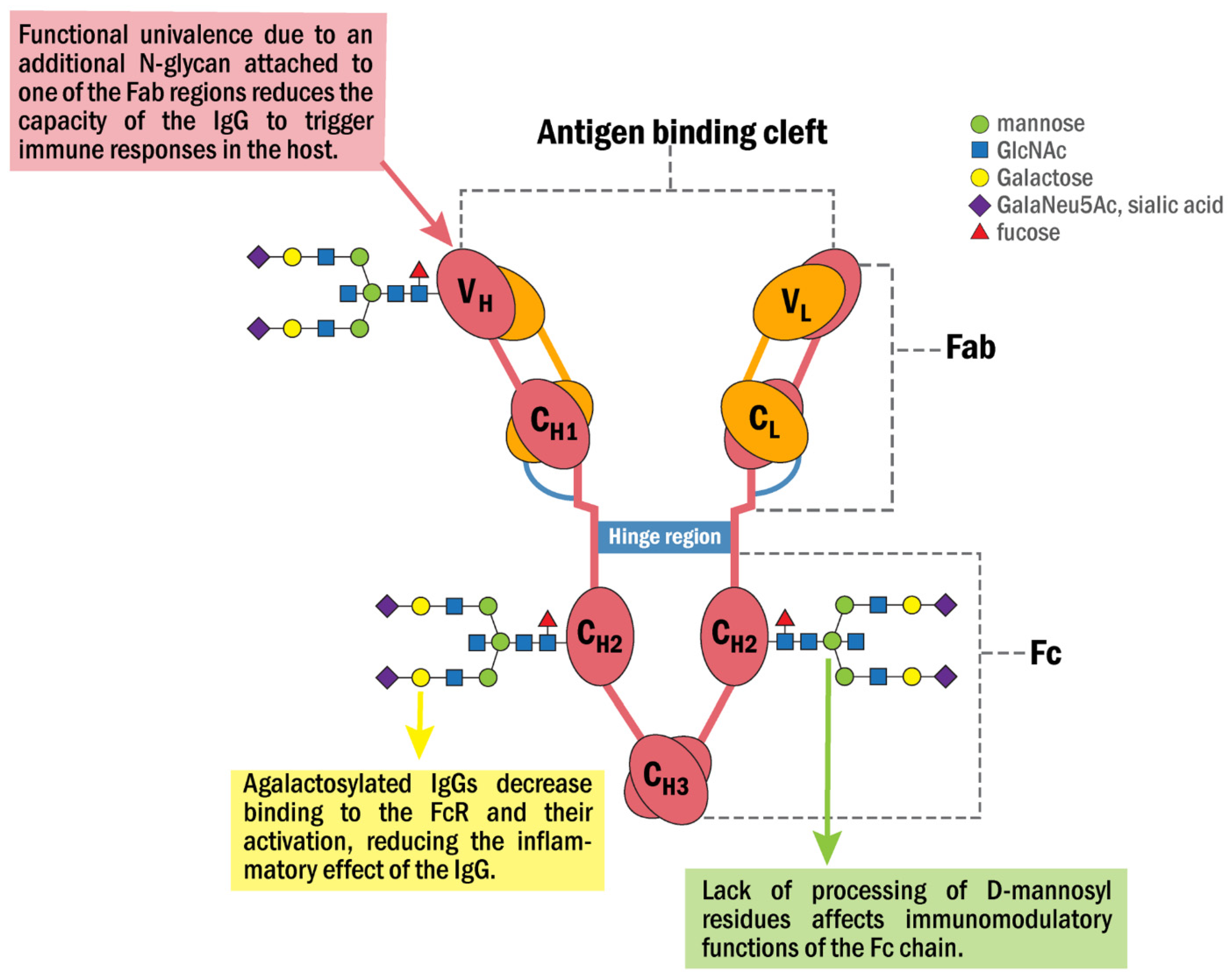
- Van de Boyenkamp, F.S.; Hafkenscheid, L.; Rispens, T.; Rombouts, Y. The emerging importance of IgG Fab glycosylation in immunity. J. Immunol. 2016, 196, 1435–1441.
- Wright, A.; Morrison, S.L. Antibody variable region glycosylation: Biochemical and clinical effects. Springer Semin. Immunopathol. 1993, 15, 259–273.
- Russel, A.; Adua, E.; Ugrina, I.; Laws, S.; Wang, W. Unravelling immunoglobulin G Fc N-glycosylation: A dynamic marker potentiating predictive, preventive and personalized medicine. Int. J. Mol. Sci. 2018, 19, 390.
- Lee, D.M.; Weinblatt, M.E. Rheumatoid arthritis. Lancet 2001, 358, 903–911.
- Stevens, C.R.; Williams, R.B.; Farrell, A.J.; Blake, D.R. Hypoxia and inflammatory synovitis: Observations and speculation. Ann. Rheum. Dis. 1991, 50, 124–132.
- Yoo, S.-A.; You, S.; Yoon, H.-J.; Kim, D.-H.; Kim, H.-S.; Lee, K.; Ahn, J.H.; Hwang, D.; Lee, A.S.; Kim, K.-J.; et al. A novel pathogenic role of ER chaperone GRP78/BiP in rheumatoid arthritis. J. Exp. Med. 2012, 209, 871–886.
- Masson-Bessière, C.; Sabbag, M.; Durieux, J.J.; Nogueira, L.; Vincent, C.; Girbal-Neuhausere, E.; Durroux, R.; Cantagrel, A.; Serre, G. In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin. Exp. Immunol. 2000, 119, 544–552.
- Kim, H.R. Anti-citrullinated antibodies in rheumatoid arthritis: A bridge between genetic predisposition and autoimmunity. Korean J. Intern. Med. 2013, 28, 25–28.
- Yamada, R. Peptidylarginine deiminase type 4, anticitrullinated peptide antibodies, and rheumatoid arthritis. Autoimmun. Rev. 2005, 4, 201–206.
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402.
- Shoda, H.; Fujio, K.; Shibuya, M.; Okamura, T.; Sumitomo, S.; Okamoto, A.; Sawada, T.; Yamamoto, K. Detection of autoantibodies to citrullinated BiP in rheumatoid arthritis patients and pro-inflammatory role of citrullinated BiP in collagen-induced arthritis. Arthritis. Res. Ther. 2011, 13, R191.
- Rombouts, Y.; Willemze, A.; van Beers, J.B.C.; Shi, J.; Kerkman, P.F.; van Toorn, L.; Janssen, G.M.C.; Zaldumbide, A.; Hoeben, R.C.; Pruijn, G.J.M.; et al. Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann. Rheum. Dis. 2016, 75, 578–585.
- Van Zeben, D.; Rook, G.A.; Hazes, J.M.; Zwinderman, A.H.; Zhang, Y.; Ghelani, S.; Rademacher, T.W.; Breedveld, F.C. Early agalactosylation of Igg is associated with a more progressive disease course in patients with rheumatoid arthritis: Results of a follow-up study. Rheumatology 1994, 33, 36–43.
- Rombouts, Y.; Ewing, E.; van de Stadt, L.A.; Selman, M.H.J.; Trouw, L.A.; Deelder, A.M.; Huizinga, T.W.J.; Wuhrer, M.; Van Schaardenburg, D.; Toes, R.; et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 234–241.
- Ercan, A.; Cui, J.; Chatterton, D.E.W.; Deane, K.D.; Hazen, M.M.; Brintnell, W.; O’Donnell, C.I.; Derber, L.A.; Weinblatt, M.E.; Shadick, N.A.; et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2239–2248.
- Aghamollaei, H.; Gargari, S.L.M.; Ghanei, M.; Rasaee, M.J.; Amani, J.; Bakherad, H.; Famoosh, G. Structure prediction, expression and antigenicity of c-terminal of GRP78. Biochem. Mol. Biol. Inc. 2017, 64, 117–126.
- Lu, M.C.; Lai, N.S.; Yu, H.C.; Huang, H.B.; Hsieh, S.C.; Yu, C.L. Anti-citrullinated protein antibodies bind surface-expressed citrullinated grp78 on monocyte-macrophages and stimulate tumor necrosis factor alpha production. Arthritis Rheum. 2010, 62, 1213–1223.
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK antigen-activated protein kinases via binding to surface-expressed citrullinated GRP78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566.
- Gharesi-Fard, B.; Zare, M.; Kamali-Sarvestani, E. The reaction of placerntal GRP78 protein with sera from women with multiple sclerosis. Iran. J. Immunol. 2017, 14, 306–315.
- Matsueda, Y.; Arinuma, Y.; Nagai, T.; Hirohata, S. Elevation of sereum anti-glucose-regulated protein 78 antibodies in neuropsychiatric systemic lupus erythematosus. Lupus Sci. Med. 2018, 5, e000281.
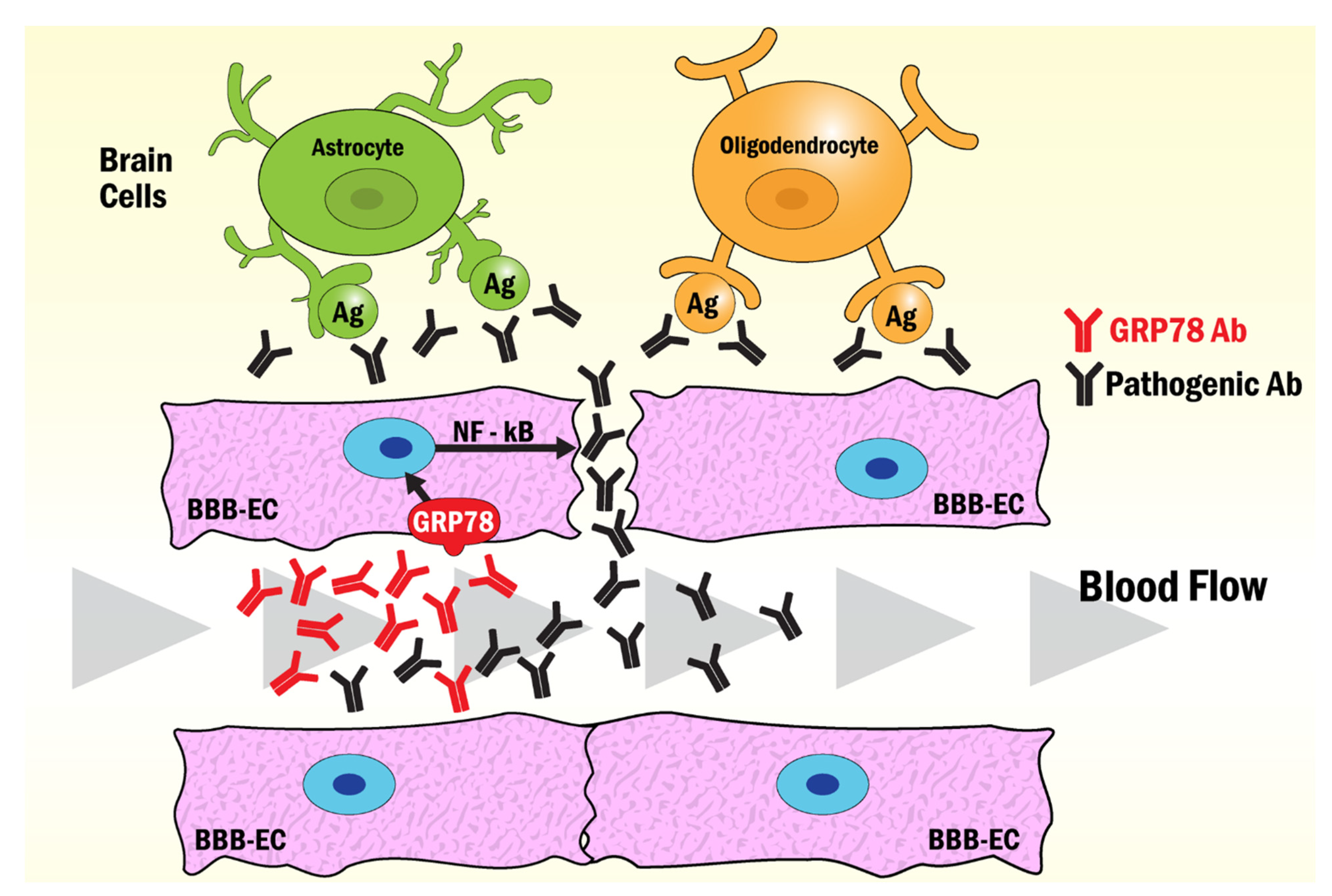
- Shimizu, F.; Ogawa, R.; Mizukami, Y.; Watanabe, K.; Hara, K.; Kadono, C.; Takahashi, T.; Misu, T.; Takeshita, Y.; Sano, Y.; et al. GRP78 antibodies are associated with blood-brain barrier breakdown in anti-myelin oligodendrocyte glycoprotein antibody-associated disorder. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1038.
- Shimizu, F.; Schaller, K.L.; Owens, G.P.; Clotleur, A.C.; Kellner, D.; Takeshita, Y.; Obermeier, B.; Kryzer, T.J.; Sano, Y.; Kanda, T.; et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci. Transl. Med. 2017, 9, eaai9111.
- Noseworthy, F.D.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952.
- Wuhrer, M.; Selman, M.H.; McDonnell, L.A.; Kümpfel, T.; Derfuss, T.; Khademi, M.; Olsson, T.; Hohlfeld, R.; Meinl, E.; Krumbholz, M. Pro-inflammatory pattern of IgG1 Fc glycosylation in multiple sclerosis cerebrospinal fluid. J. Neuroinflamm. 2015, 12, 235.
- Liang, M.H.; Corzillius, M.; Bae, S.C.; Lew, R.A.; Fortin, P.R.; Gordon, C.; Isenberg, D.; Alarcón, G.S.; Straaton, K.V.; Denburg, J. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999, 42, 599–608.
- Vučković, F.; Krištić, J.; Gudelj, I.; Teruel, M.; Keser, T.; Pezer, M.; Pučić-Baković, M.; Štambuk, J.; Trbojević-Akmačić, I.; Barrios, C.; et al. Association of Systemic Lupus Erythematosus With Decreased Immunosuppressive Potential of the IgG Glycome. Arthritis Rheumatol. 2015, 67, 2978–2989.
- Titulaer, M.J.; Lang, B.; Verschuuren, J.J. Lambert-Eaton myathenic syndrome from clinical characteristics to therapeutic strategies. Lancet 2011, 10, 1098–1107.
- Jarius, S.; Wildemann, B. Thed history of neuromyelitis optica. J. Neuroinflamm. 2013, 10, 8.
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477.
- Waters, P.J.; McKeon, A.; Leite, M.I.; Rajasekharan, S.; Lennon, V.A.; Villalobos, A.; Palace, J.; Mandrekar, J.N.; Vincent, A.; Bar-Or, A.; et al. Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology 2012, 78, 665–671.
- Roemer, S.F.; Parisi, J.E.; Lennon, V.A.; Benarroch, E.E.; Lassmann, H.; Bruck, W.; Mandler, R.N.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007, 10, 1194–1205.
- Shimizu, F.; Sano, Y.; Takahashi, T.; Haruki, H.; Saito, K.; Koga, M.; Kanda, T. Sera from neuromyelitis optica patients disrupt the blood-brain barrier. J. Neurol. Neurosurg. Psychiatry 2012, 83, 288–297.
- Renaudineau, Y.; Duguè, C.; Dueymes, M.; Youinou, P. Antiendothelial cell antibodies in systemic lupus erythematosus. Autoimmun. Rev. 2002, 1, 365–372.
- Yu, Y.J.; Watts, R.J. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 2013, 10, 459–472.

