2. Molecular Mechanisms and ER Stress Conditions That Promote GRP78 Autoimmunity in Cancer
GRP78 binds hydrophobic surfaces on newly synthesized polypeptides and is first in line for protein folding, a function that is enhanced when misfolded polypeptides accumulate within the ER as a consequence of cellular stress
[2][15][2,15]. GRP78 binds to unfolded proteins in its ATP-bound form and mediates their folding at the expense of ATP
[16]. When GRP78 functions as a chaperone, it dissociates from the ER transmembrane stress sensor proteins inositol requiring enzyme 1 (Ire1), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6), which trigger the UPR
[17]. Once activated, the UPR signaling promotes the transcription of numerous chaperones to protect the cell from accumulated proteins, and GRP78 itself is a transcriptional target of the UPR via ER stress-responsive elements that can bind to ATF6
[18]. The UPR leads to elevated expression of GRP78 in tumor tissue and controls not only its aberrant localization in the cytosol and the mitochondria but also in the plasma membrane
[19]. The over-expression of GRP78 leads to the saturation of the KDEL receptor retrieval mechanism
[20], permitting evasion and migration of GRP78 from the ER to these aberrant sites, a phenomenon common to several types of cancer
[21]. In prostate cancer and melanoma, csGRP78 induces the production of GRP78 autoantibodies
[22][23][22,23]. The interaction of these autoantibodies with csGRP78 triggers phosphoinositide 3-kinase (PI3K), protein kinase B (Akt) and mitogen-activated protein kinase (MAPK) signaling pathways, increasing cell survival during tumor growth and metastasis
[24][25][24,25]. GRP78 is also functionally linked to the immune system via binding to the major histocompatibility complex class I (MHC-I), regulating peptide presentation by MHC-I in the plasma membrane
[26]. Overexpression of csGRP78 under ER stress decreases MHC-I expression on the surface
[27], and this mechanism is used by tumor cells to evade immune surveillance
[28][29][28,29].
In prostate cancer, the GRP78 autoantibody recognizes a tertiary structure motif with the amino acid sequence CNVKSDKC
[30], contained within the GRP78 primary amino acid sequence L
98IGRTWNDPSVQQDIKFL
115 (L
98-L
115)
[22]. This csGRP78 N-terminal region site is also a second receptor for activated α
2-macroglobulin (α
2M*)
[31] which initiates pro-proliferative signaling pathways upon the binding of either α
2M* or GRP78 autoantibodies to prostate
[22] or melanoma cancer cells
[23]. Structural analyses of anti-GRP78 antibodies from a group of patients at different stages of malignant melanoma show that the IgG Fab is asymmetrically glycosylated, while the Fc regions are aberrantly glycosylated
[23] in a manner similar to that observed in ovarian cancer
[32]. Functionally, the asymmetric glycosylation matters, as reflected by the capacity of these IgG to activate the Akt signaling pathway while promoting the growth of prostate, lung and gastric cancers
[33][34][33,34]. Asymmetric antibodies have two paratopes, one of high affinity, with K
d similar to that of symmetric antibodies and the other one with an affinity for the antigen significantly lower
[35]. As a consequence, these IgG function as univalent and fail to trigger some of the major biological reactions of the immune response such as complement fixation, phagocytic activity and antigen clearance, thereby potentially exerting protective immunomodulatory effects on the host tissues
[36]. The aberrant glycosylation of the Fc region in the GRP78 Abs in malignant melanoma
[23] is the result of a lack of processing of D-mannosyl residues, which leads to a change in the size of the IgG-Fc fragment (27–33 kDa)
[37]. These changes decrease their affinity for the Fc receptor (FcR), inducing changes in immune modulation that affect FcR activation
[38]. These observations suggest that anti-GRP78 antibodies produced in cancer patients are functionally synthesized to protect the target tissue from immune responses, providing the tumor with defensive mechanisms that facilitate cell survival and proliferation.
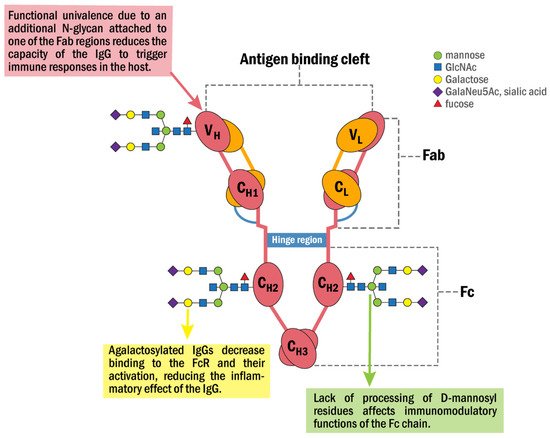
Figure 1 shows a model summarizing some of the properties and functions that characterize the anti-GRP78 IgG and its associated N-glycan structures identified in cancer.
Figure 1. Schematic representation of the anti-GRP78 IgG molecular structure and its associated N-glycan structures identified in cancer patients. It consists of two interconnected heavy (H) and two light chains (L) with two domains that infer different properties, the fragment antigen-binding (Fab) and fragment crystallizable (Fc) domains. The Fab domain is responsible for recognizing and binding the antigen. The Fc domain contains two glycans attached to Asn297 in conserved regions of the CH2 domain. The Fc domain binds to the FcR on natural killers and other inflammatory cells. In cancer, the Fab region is asymmetrically glycosylated with one additional N-glycan that converts the anti-GRP78 IgG into a univalent molecule that binds the antigen with a decreased affinity, preventing it from triggering some IgG-linked functions of the immune response such as complement fixation, phagocytic activity and antigen clearance. Moreover, the N-glycans of the Fc region are aberrantly glycosylated, mainly at the levels of D-mannosylation and galactosylation with a reduction in the affinity of the antibody for the FcR and its activation, thus inhibiting the inflammatory effects of the IgG.
3. Mechanisms and ER Stress Conditions That Promote GRP78 Autoimmunity in RA
RA is a systemic autoimmune disease characterized by chronic inflammation of the joints resulting in hypertrophy of the synovium called pannus, which ultimately leads to joint destruction
[39]. The ER response has been implicated in chronic autoimmune inflammatory diseases
[12] and ER stressors such as hypoxia, low glucose, and the pro-inflammatory cytokine milieu are responsible for the abnormal proliferation of RA fibroblast-like synoviocytes (RA-FLS) and angiogenesis, promoting pannus formation
[40]. During these processes, down-regulation of GRP78 increases apoptosis of RA-FLS, whereas its overexpression prevents R-FLS from apoptotic death induced by ER-stressors
[41]. The mechanisms operating in the UPR response of RA cells are similar to those described above for cancer cells
[14]; however, during the progress of RA, B cells differentiate into plasma cells and infiltrate the synovial membrane where they synthesize autoantibodies such as immunoglobulin M rheumatoid factor (RF) and anti-cyclic citrullinated peptide antibodies (ACPA)
[42][43][42,43].
Citrullination is a posttranslational modification of proteins catalyzed by peptidylarginine deiminase (PADI) in the presence of a high concentration of calcium that causes a loss of basic charge(s), which can influence the protein structure and create new epitopes recognized by the immune system
[44]. In RA patients, the PADI type 4 (PADI4) gene in hematological and synovial tissue is a susceptibility locus for RA that affects the stability of transcripts and is associated with levels of antibody to citrullinated peptides in their sera. The PADI4 haplotype associated with susceptibility to RA increases the production of citrullinated peptides acting as autoantigens in some individuals, leading to a heightened risk of developing the disease, suggesting that genetic factors could cause RA
[45]. As a target of PADI4, the citrullinated GRP78 (citGRP78) is arthritogenic in RA mice models, leading to the generation of ACPA which increases the severity of the disease
[46].
Although the structure of the anti-citGRP78 IgG was not specifically studied in RA, ACPA-IgG molecules show a 10–20 kDa higher molecular weight compared with non-autoreactive IgG, resulting from Fab-linked N-glycosylation that decreases the binding avidity of ACPA for citrullinated antigens
[47]. The lower target affinity induced by N-glycosylation of the RA ACPA-IgGs suggests a lower pathogenicity for these antibodies; however, the Fc-linked aberrant decrease in galactosylation and increase in fucosylation patterns observed in RA patients further increases a more pro-inflammatory phenotype of these antibodies
[48][49][48,49]. The integrity of the IgG Fc region oligosaccharide chain is necessary for the activation of the complement, interactions with the FcR, and antibody-dependent cell-mediated cytotoxicity, all of which are perturbed by truncation of the IgG Asn
297 oligosaccharide, where hypogalactosylation is a significant factor in the pathogenesis of RA
[50].
The epitope specificity of the anti-citGRP78 antibodies in RA was identified using citrullinated peptides in which the arginine residues were replaced with citrulline residues. The RA patient serum antibody levels to GRP78
279–298 R289citrulline (P12), GRP78
295–314 R305 citrulline (P15) and GRP78
500–519 R510citrulline (P23) were significantly increased compared with those of native peptides in RA patients, demonstrating their significance for antibody recognition in RA, in particular the epitope contained in peptide P23 in the GRP78 C-terminal region
[41]. The high immunogenicity of the GRP78 C-terminal region was further confirmed in a separate study using a cloned GRP78
409–653 C-terminal segment, structurally similar to that in the whole protein
[51]. The anti-citGRP78 antibodies bind to csGRP78 on monocytes and activate extracellular signal-regulated protein kinases (ERK) and Jun N-terminal Kinase (JNK) pathways, leading to activation of nuclear factor kappa b (NF-κB) and production of tumor necrosis factor alpha (TNF-α)
[52][53][52,53]. The production of TNF-α further increases csGRP78 expression in RA-FLS, leading to cell apoptosis
[14].
4. GRP78 Autoantibodies in Immune-Mediated Neurological Diseases
GRP78 autoantibodies have been identified in MS
[54], NPSLE
[55], AMOGAD
[56], LEMS
[8], and NMO
[57]. MS is a chronic inflammatory demyelinating disease of the central nervous system (CNS), characterized by the presence of focal demyelinated plaques within the white matter
[58]. Although the structure of the anti-GRP78 IgG has not been specifically analyzed, the Fc region in IgG1 from MS patients shows reduced fucosylation and galactosylation, and increased bisecting GlcNAc, that promotes a higher pro-inflammatory activity of this immunoglobulin
[59].
NPSLE is a complication of SLE characterized by serious cognitive defects which fluctuate over time such as difficulties in attention, concentration and memory
[60]. Patients with NPSLE show significantly elevated serum levels of anti-GRP78 antibodies compared with healthy controls
[55]. Although the structure of the anti-GRP78 IgG has not been specifically analyzed, significant differences in the IgG glycosylation between SLE patients and controls have been reported
[61]. These changes include decreases in the galactosylation and sialylation, which regulate proinflammatory and anti-inflammatory actions of IgG, as well as decreased core fucose and increased bisecting N-acetylglucosamine, which affect antibody-dependent cell-mediated cytotoxicity
[61], thereby suggesting that aberrant IgG glycosylation of the anti-GRP78 antibody may be an important pathological mechanism in SLE.
AMOGAD was recently recognized as a new pathology in the spectrum of inflammatory diseases, which differs from either MS or NMO. The serum levels of anti-GRP78 antibodies in patients with AMOGAD are significantly higher than those of MS patients or healthy controls
[56]. LEMS is an autoimmune disease of the neuromuscular junction commonly defined as a paraneoplastic neurological syndrome, involving muscle weakness, areflexia and autonomic dysfunction
[62]. Serum levels of anti-GRP78 antibodies of LEMS patients are high, and none were detected in the control subjects
[8].
NMO is a severe inflammatory autoimmune disorder of the CNS that affects both adults and children. NMO was historically considered a variant of MS
[63]; however, the discovery and identification of an NMO IgG autoantibody, specific for aquaporin-4 water channel (AQP4-Ab)
[64], facilitated clinical diagnosis and early treatment of NMO
[65]. Patient tissue immunopathology demonstrates that astrocytes are the principal cell target in NMO
[66]. The circulating AQP4-Abs move across the blood-brain barrier (BBB), reaching brain astrocytes after leakage induced by anti-GRP78 antibodies also present in the circulation of NMO patients
[67]. The mechanism by which the anti-GRP78 antibodies break the permeability of the BBB involves their binding to the scGRP78 on the brain microvascular endothelial cells (BMECs), inducing nuclear translocation of NF-κB that facilitates ICAM-1 transcription and promotes binding of activated immune cells that enhance the diameter of cerebral blood vessels in
[58]. Similar mechanisms have been observed with anti-GRP78 antibodies in AMOGAD
[56] and LEMS
[8] that induce brain endothelial cells to open the BBB, thereby allowing access to other pathogenic antibodies in the CNS. A compromised BBB and antiendothelial cell antibodies have been found in patients with SLE
[68], suggesting that csGRP78 in brain endothelial cells may also be a target of antiGRP78 antibodies in SLE
[69]. The mechanism of disruption of the BBB by GRP78 autoantibodies in NMO, AMOGAD and SLE is summarized in
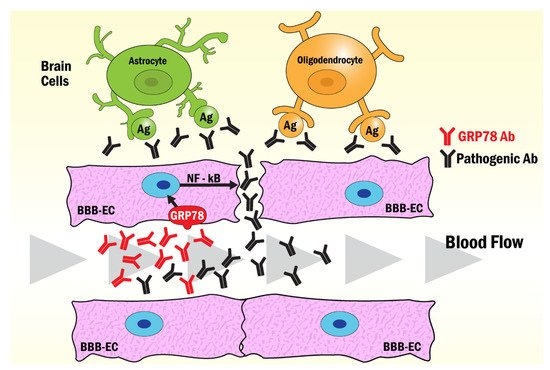
Figure 2.
Figure 2. Model of the mechanism of BBB opening induced by GRP78 antibodies common to NMO, AMOGAD, LEMS and SLE. The binding of antibodies to GRP78 on the surface of BBB-endothelial cells induces the activation of NF-κB signaling pathways that disrupt the tight junction enhancing the diffusion of pathogenic antibodies into the brain. Astrocytes react with AQP4-Abs in NMO, while oligodendrocytes react with pathogenic anti-myelin Abs in AMOGAD. Pathogenic antibodies to brain antigens found in LEMS and SLE may also function through this mechanism to induce the damage observed in these pathologies.