Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rainer Ullrich Pliquett | -- | 1189 | 2022-06-09 07:38:17 | | | |
| 2 | Sirius Huang | Meta information modification | 1189 | 2022-06-09 07:42:13 | | | | |
| 3 | Sirius Huang | Meta information modification | 1189 | 2022-06-10 03:49:02 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Pliquett, R. An Updated Classification of Cardiorenal Syndrome. Encyclopedia. Available online: https://encyclopedia.pub/entry/23864 (accessed on 24 July 2026).
Pliquett R. An Updated Classification of Cardiorenal Syndrome. Encyclopedia. Available at: https://encyclopedia.pub/entry/23864. Accessed July 24, 2026.
Pliquett, Rainer. "An Updated Classification of Cardiorenal Syndrome" Encyclopedia, https://encyclopedia.pub/entry/23864 (accessed July 24, 2026).
Pliquett, R. (2022, June 09). An Updated Classification of Cardiorenal Syndrome. In Encyclopedia. https://encyclopedia.pub/entry/23864
Pliquett, Rainer. "An Updated Classification of Cardiorenal Syndrome." Encyclopedia. Web. 09 June, 2022.
Copy Citation
Cardiorenal syndrome (CRS) is defined as progressive, combined cardiac and renal dysfunction. Here, the pathomechanisms and clinical hallmarks of both chronic heart failure and chronic kidney disease are presented, and an updated classification of CRS is proposed.
cardiorenal syndrome
chronic heart failure
chronic kidney disease
acute kidney injury
acute heart failure
1. Clinical and Physiological Hallmarks of Cardiorenal Syndrome
Besides chronic heart failure (CHF), the activation of the sympathetic nervous system (SNS) [1] and chronically elevated inflammatory serum parameters [2] have been identified as clinical hallmarks of chronic kidney disease (CKD). Both sympathoactivation [3][4] and systemic inflammation [5] are considered to be key pathomechanisms in cardiorenal syndrome (CRS) as well. Hemodynamic abnormalities in CRS include venous congestion due to increased right ventricular filling pressure accompanied by a tricuspid annular dilatation with ensuing regurgitation [6]. In the latter pathomechanism, the centrally venous congestion impedes the renal venous blood flow and favors an intrarenal edema, which may further impede the intrarenal arterial perfusion. As proof, kidney sonography may demonstrate an attenuated arterial intrarenal perfusion being absent in the periphery of the kidneys. Clearly, in CRS, an arterial underfilling mechanism due to renal hypoperfusion, e.g., in heart failure with reduced ejection fraction and/or during hypotension, may represent an alternative explanation as to why renal function may cease in CRS. In fact, venous congestion due to increased right ventricular filling pressures, e.g., in diastolic left ventricular hypertrophy with left ventricular diastolic dysfunction, and renal hypoperfusion during hypotension or during periods of cardiac decompensation may lead to acute kidney injury with or without a pre-existing chronic kidney disease in CRS. As a proof of concept, once renal venous congestion alleviates—e.g., following the paracentesis of ascites—renal function improves readily [7]. Likewise, when renal venous congestion is diagnosed in clinical medicine, the use of loop diuretics regularly improves both venous congestion and renal function. Point-of-care ultrasound of abdominal veins including portal-vein flow pattern and inferior vena cava size [8], the sonographic detection of extravascular lung fluid [9], and the determination of Doppler-sonographic intrarenal venous flow patterns [10] may help direct therapy to achieve less venous congestion and an augmented renal function in hypervolemic CRS. Euvolemia, or a steady state with the least possible venous congestion, is to be maintained by achieving a low maintenance dose of loop diuretics by adjusting the recommended daily intake of water and by lowering salt intake to 2–3 g per day. Once renal dysfunction due to either venous congestion or renal arterial hypoperfusion occurs, cardiac function may deteriorate. Conversely, once renal function resumes, cardiac tricuspid annular dilatation may improve due to less right ventricular filling pressure. As a caveat, accidental hypovolemia due to polyuria during renal recovery, hyperglycemia-related polyuria in diabetes mellitus, or a prolonged use of high-dose loop-diuretics may lead to hypovolemic shock with attenuated renal perfusion and/or to acute cardiac failure. In CRS, due to valvular heart disease, because of both signs and symptoms, hemodynamics and therapeutic implications may differ. Therefore, a differentiation between valvular and nonvalvular CRS is needed, if therapy stratification is the goal. Besides changes in hemodynamics, both anemia [11] and systemic inflammation [5] represent unifying final pathomechanisms in CRS.
2. Signs and Symptoms of Underlying Chronic Kidney Disease and Chronic Heart Failure
As outlined in Table 1, both CHF and advanced CKD share several signs, symptoms, and laboratory key findings. Based on the clinical exam, it is a challenge to determine the underlying etiology of CRS—e.g., whether acute or chronic heart failure preceded an acute or chronic kidney injury. Peripheral edema, pulmonary venous congestion, and interstitial pulmonary edema are commonly detected in advanced CKD, in CHF, or in the combination thereof, CRS. As for the pathomechanism, an activated SNS is regularly found both in advanced CKD [1] and in CHF [12]. SNS activation is governed by the afferent loop of cardiovascular reflexes and by the brain renin–angiotensin system [13]. The renin–angiotensin system in the brain, the renin–angiotensin–aldosterone system (RAAS) in the periphery, and the SNS are all interrelated: plasma angiotensin 2 may contribute to hypothalamic SNS activation at anatomic loci where a tight brain–blood barrier is lacking [14]. Likewise, an activated SNS may lead to renin release from juxtaglomerular cells via the sympathetic renal nerves [15]. In CKD, renin activation leading to RAAS activation is a hallmark [16]. In addition, hyperkalemia independently leads to aldosterone activation in advanced CKD. Conversely, the treatment of hyperkalemia using the potassium binder Patiromer has the potential to lower plasma aldosterone in advanced CKD [17]. Underlying sarcopenia or protein-energy wasting has been recognized for both CHF and ESRD. Hypoalbuminemia, which is regularly found in both CHF [18] and end-stage renal disease (ESRD) [19] is associated with an increased mortality. As for ESRD, nutritional efforts were unable to prevent the prevalent catabolism [20]. The paradoxical association between low levels of low-density lipoprotein cholesterol and mortality has been described both in CHF [21] and ESRD [22]. Even though a causality was not proven, the pathomechanism may relate to an impaired liver function comprising an impaired biosynthesis of transport proteins both in CHF and in CRS, as 47% of ESRD patients included in this prospective observational study had CHF comorbidity [22]. Lastly, anemia has associations with both CHF [23] and CKD [24]. While a decreased erythropoietin production occurs in advanced CKD [25], both early stages of CHF [26] and advanced CKD [27] are associated with iron deficiency due to increased hepcidin plasma levels down-regulating enteral iron absorption. Systemic inflammation represents a candidate pathomechanism for hepcidin activation in both CHF [28] and advanced CKD [2].
Table 1. Clinical characteristics of chronic kidney disease and chronic heart failure.
| Chronic Kidney Disease (KDIGO G4-G5nonD) |
Chronic Heart Failure (NYHA III-IV) | |
|---|---|---|
| Peripheral edema | + | + |
| Pulmonary venous congestion | (+) | + |
| Interstital pulmonary edema | (+) | (+) |
| Sympathoactivation | + | + |
| Renin–angiotensin–aldosterone activation | + | + |
| Hypoalbuminemia | (+) | (+) |
| Cholesterol paradox | + | + |
| Anemia | + | + |
| Microinflammation | + | + |
3. Updated Classification of Cardiorenal Syndrome
In retrospect, the older classification differentiating between severe and non-severe CRS [29] conveyed prognostic information when CRS is coined “severe”. For clinicians, this differentiation implies that immediate action is needed to avoid a vicious cycle towards progressive cardiac and renal failure and death. In contrast, the current consensus classification of CRS [30] highlights the complexity of the underlying causes; however, it lacks therapeutic or prognostic implications. Here, the proposed update of the consensus classification of CRS specifies whether CRS is acute or chronic, whether a valvular or nonvalvular heart disease is present and whether CRS associates with hyper- or hypovolemia (Figure 1). The first point, the descriptive information on an acute or chronic course of disease, rather simplifies the current consensus classification. CRS types I, III, and V may be regarded as acute CRS and types II, IV, and V as chronic CRS. Etiologic information on valvular and nonvalvular heart disease is necessary to direct therapeutic decisions. Valvular heart disease may be subjected to a correctional cardiologic procedure or to cardiac surgery. Conversely, nonvalvular heart disease as a component of CRS may be subjected to evidence-based medical therapies. Lastly, if euvolemia is absent, information on a prevalent hyper- or hypovolemia needs to be provided to further direct medical therapy.
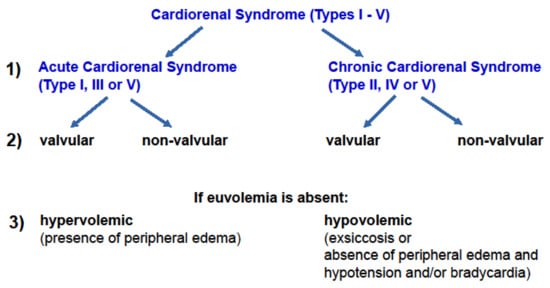
Figure 1. A proposed new classification of cardiorenal syndrome. An upgraded classification of CRS requires clinical information that further specifies current consensus classification of CRS [30]. Aside from etiology and time course of CRS, clinical hallmarks such as the volemic state are considered.
References
- Schlaich, M.P.; Socratous, F.; Hennebry, S.; Eikelis, N.; Lambert, E.A.; Straznicky, N.; Esler, M.D.; Lambert, G.W. Sympathetic activation in chronic renal failure. J. Am. Soc. Nephrol. 2009, 20, 933–939.
- Akchurin, O.M.; Kaskel, F. Update on inflammation in chronic kidney disease. Blood Purif. 2015, 39, 84–92.
- Virzì, G.M.; Clementi, A.; Brocca, A.; de Cal, M.; Vescovo, G.; Granata, A.; Ronco, C. The hemodynamic and nonhemodynamic crosstalk in cardiorenal syndrome type 1. Cardiorenal Med. 2014, 4, 103–112.
- Clementi, A.; Virzì, G.M.; Brocca, A.; de Cal, M.; Pastori, S.; Clementi, M.; Granata, A.; Vescovo, G.; Ronco, C. Advances in the pathogenesis of cardiorenal syndrome type 3. Oxid. Med. Cell. Longev. 2015, 2015, 148082.
- Linhart, C.; Ulrich, C.; Greinert, D.; Dambeck, S.; Wienke, A.; Girndt, M.; Pliquett, R.U. Systemic inflammation in acute cardiorenal syndrome: An observational pilot study. ESC Heart Fail. 2018, 5, 920–930.
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.H.W. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 2009, 53, 589–596.
- Mullens, W.; Abrahams, Z.; Skouri, H.N.; Francis, G.S.; Taylor, D.O.; Starling, R.C.; Paganini, E.; Tang, W.H. Elevated intra-abdominal pressure in acute decompensated heart failure: A potential contributor to worsening renal function? J. Am. Coll. Cardiol. 2008, 51, 300–306.
- Argaiz, E.R.; Rola, P.; Gamba, G. Dynamic Changes in Portal Vein Flow during Decongestion in Patients with Heart Failure and Cardio-Renal Syndrome: A POCUS Case Series. Cardiorenal Med. 2021, 11, 59–66.
- Pellicori, P.; Platz, E.; Dauw, J.; Ter Maaten, J.M.; Martens, P.; Pivetta, E.; Cleland, J.; McMurray, J.; Mullens, W.; Solomon, S.D.; et al. Ultrasound imaging of congestion in heart failure: Examinations beyond the heart. Eur. J. Heart Fail. 2021, 23, 703–712.
- Husain-Syed, F.; Birk, H.W.; Ronco, C.; Schörmann, T.; Tello, K.; Richter, M.J.; Wilhelm, J.; Sommer, N.; Steyerberg, E.; Bauer, P.; et al. Doppler-Derived Renal Venous Stasis Index in the Prognosis of Right Heart Failure. J. Am. Heart Assoc. 2019, 8, e013584.
- Pallangyo, P.; Fredrick, F.; Bhalia, S.; Nicholaus, P.; Kisenge, P.; Mtinangi, B.; Janabi, M.; Humphrey, S. Cardiorenal Anemia Syndrome and Survival among Heart Failure Patients in Tanzania: A Prospective Cohort Study. BMC Cardiovasc. Disord. 2017, 17, 59.
- Grassi, G.; Mancia, G.; Esler, M. Central and Peripheral Sympathetic Activation in Heart Failure. Cardiovasc. Res. 2021, cvab222.
- Zucker, I.H.; Xiao, L.; Haack, K.K.V. The Central RAS and Sympathetic Nerve Activity in Chronic Heart Failure. Clin. Sci. 2014, 126, 695–706.
- Biancardi, V.C.; Stern, J.E. Compromised blood-brain barrier permeability: Novel mechanism by which circulating angiotensin II signals to sympathoexcitatory centres during hypertension. J. Physiol. 2016, 594, 1591–1600.
- Schweda, F.; Friis, U.; Wagner, C.; Skott, O.; Kurtz, A. Renin release. Physiology 2007, 22, 310–319.
- Siragy, H.M.; Carey, R.M. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. Am. J. Nephrol. 2010, 31, 541–550.
- Weir, M.R.; Bakris, G.L.; Gross, C.; Mayo, M.R.; Garza, D.; Stasiv, Y.; Yuan, J.; Berman, L.; Williams, G.H. Treatment with patiromer decreases aldosterone in patients with chronic kidney disease and hyperkalemia on renin-angiotensin system inhibitors. Kidney Int. 2016, 90, 696–704.
- Horwich, T.B.; Kalantar-Zadeh, K.; MacLellan, R.W.; Fonarow, G.C. Albumin levels predict survival in patients with systolic heart failure. Am. Heart J. 2008, 155, 883–889.
- Alves, F.C.; Sun, J.; Qureshi, A.R.; Dai, L.; Snaedal, S.; Bárány, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P. The higher mortality associated with low serum albumin is dependent on systemic inflammation in end-stage kidney disease. PLoS ONE 2018, 13, e0190410.
- Zilles, M.; Betz, C.; Jung, O.; Gauer, S.; Hammerstingl, R.; Wächtershäuser, A.; Vogl, T.J.; Geiger, H.; Asbe-Vollkopf, A.; Pliquett, R.U. How to Prevent Renal Cachexia? A Clinical Randomized Pilot Study Testing Oral Supplemental Nutrition in Hemodialysis Patients with and Without Human Immunodeficiency Virus Infection. J. Ren. Nutr. 2018, 28, 37–44.
- Rauchhaus, M.; Clark, A.L.; Doehner, W.; Davos, C.; Bolger, A.; Sharma, R.; Coats, A.J.; Anker, S.D. The relationship between cholesterol and survival in patients with chronic heart failure. J. Am. Coll. Cardiol. 2003, 42, 1933–1940.
- Liu, Y.; Coresh, J.; Eustace, J.A.; Longenecker, J.C.; Jaar, B.; Fink, N.E.; Tracy, R.P.; Powe, N.R.; Klag, M.J. Association between cholesterol level and mortality in dialysis patients: Role of inflammation and malnutrition. JAMA 2004, 291, 451–459.
- Anand, I.S.; Gupta, P. Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies. Circulation 2018, 138, 80–98.
- Babitt, J.L.; Lin, H.Y. Mechanisms of anemia in CKD. J. Am. Soc. Nephrol. 2012, 23, 1631–1634.
- Hamza, E.; Metzinger, L.; Metzinger-Le Meuth, V. Uremic Toxins Affect Erythropoiesis during the Course of Chronic Kidney Disease: A Review. Cells 2020, 9, 2039.
- Jankowska, E.A.; Malyszko, J.; Ardehali, H.; Koc-Zorawska, E.; Banasiak, W.; von Haehling, S.; Macdougall, I.C.; Weiss, G.; McMurray, J.J.; Anker, S.D.; et al. Iron status in patients with chronic heart failure. Eur. Heart J. 2013, 34, 827–834.
- Tomosugi, N.; Kawabata, H.; Wakatabe, R.; Higuchi, M.; Yamaya, H.; Umehara, H.; Ishikawa, I. Detection of serum hepcidin in renal failure and inflammation by using ProteinChip System. Blood 2006, 108, 1381–1387.
- Niebauer, J.; Volk, H.D.; Kemp, M.; Dominguez, M.; Schumann, R.R.; Rauchhaus, M.; Poole-Wilson, P.A.; Coats, A.J.; Anker, S.D. Endotoxin and immune activation in chronic heart failure: A prospective cohort study. Lancet 1999, 353, 1838–1842.
- Bongartz, L.G.; Cramer, M.J.; Doevendans, P.A.; Joles, J.A.; Braam, B. The severe cardiorenal syndrome: ‘Guyton revisited’. Eur. Heart J. 2005, 26, 11–17.
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-renal syndromes: Report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J. 2010, 31, 703–711.
More
Information
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
3 times
(View History)
Update Date:
10 Jun 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No