 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Evelyn Mendoza-Torres | -- | 4240 | 2022-06-08 20:07:16 | | | |
| 2 | Vivi Li | Meta information modification | 4240 | 2022-06-09 05:52:28 | | |
Video Upload Options
Cancer is one of the main causes of death globally. Most of the molecular mechanisms underlying cancer are marked by complex aberrations that activate the critical cell-signaling pathways that play a pivotal role in cell metabolism, tumor development, cytoskeletal reorganization, and metastasis. The phosphatidylinositol 3-kinase/protein kinase-B/mammalian target of the rapamycin (PI3K/AKT/mTOR) pathway is one of the main signaling pathways involved in carcinogenesis and metastasis. Autophagy, a cellular pathway that delivers cytoplasmic components to lysosomes for degradation, plays a dual role in cancer, as either a tumor promoter or a tumor suppressor, depending on the stage of the carcinogenesis. Statins are the group of drugs of choice to lower the level of low-density lipoprotein (LDL) cholesterol in the blood. Experimental and clinical data suggest the potential of statins in the treatment of cancer. In vitro and in vivo studies have demonstrated the molecular mechanisms through which statins inhibit the proliferation and metastasis of cancer cells in different types of cancer. The anticancer properties of statins have been shown to result in the suppression of tumor growth, the induction of apoptosis, and autophagy.
1. Introduction
2. Statins
2.1. Statins and Autophagy
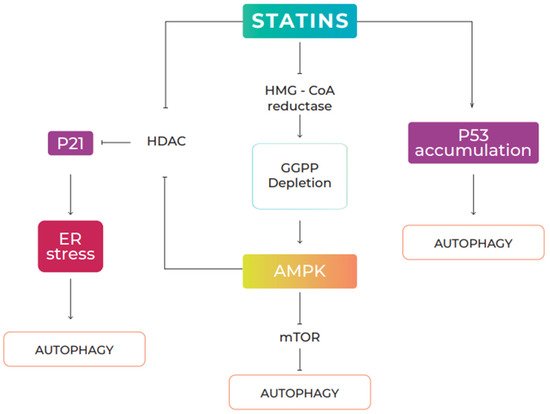
2.2. Effects of Statins on Cancer via Autophagy
| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Atorvastatin | Breast cancer | MDA-MB-231 Cells | - | 0,5, 1, 2, 4, 8 µM | Reduced the viability of cancer cells by inducing autophagy [57]. |
| Atorvastatin | Breast cancer | MCF-7 | - | 5, 10, 20, 40 y 80 μM | Decreased the proliferation of breast cancer cells through the induction of both apoptosis and autophagy [58]. |
| Atorvastatin | Ovarian cancer | Hey and SKOV3 cells | - | 1–250 μM | Inhibited the growth of ovarian cancer cell lines associated with the induction of apoptosis, autophagy, cellular stress, and G1 cell-cycle arrest [19]. |
| Atorvastatin | Cervical Cancer | SiHa and Caski Cells | Female BALB/c nude mice |
0, 5, 10, y 20, 40, 80 μM (in vitro) 50 mg/kg (in vivo) |
Reduced the viability of cervical cancer cells in vitro and in vivo by inducing apoptosis. ATO induced autophagy, and its inhibition was shown to enhance the anti-cancer effects of ATO on cervical cancer cells [60]. |
| Atorvastatin | Digestive malignancies | HCC cells (Hep3B, HepG2 and Huh7) CRC cells (HCT116 wt, HCT116 p21) |
Female BALB nude mice |
50 μM (in vitro) 50 mg/kg (in vivo) |
Inhibited cancer cell growth in vivo and in vitro by inducing apoptosis. ATO induced autophagy, and the pharmacological inhibition of autophagy was shown to enhance the anticancer effects of ATO in gastrointestinal malignancies [55]. |
| Atorvastatin | Bladder Cancer | T24 and J28 Cells | - | 0, 10, 20, 30, 40 y 50 μM | Enhanced ATP-induced apoptotic cell death in human bladder cancer cells in vitro through the pharmacological inhibition of autophagy [61]. |
| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Fluvastatin | Breast cancer | MCF-7 | - | 10 μM | Reduced cell viability through the depletion of lysosomal activities coupled with the accumulation of autophagosomes, leading to impaired autophagosome–lysosomal fusion in treated cells [62]. |
| Fluvastatin | Lung adenocarcinoma | A549 and SPC-A-1 cells | Female nude mice BALB/c | 10 μM (in vitro) 50 mg/kg (in vivo) |
Suppressed bone metastasis from lung adenocarcinoma in vivo and in vitro by triggering autophagy through the p53–AMPK-mTOR pathway [48]. |
| Fluvastatin | Lymphoma | A20 and EL4 cells | - | 0–10 μM | Induced apoptosis in lymphoma cells by activating autophagy through increased LC3-II [63]. |
| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Lovastatin | Malignant pleural mesothelioma | ACC-MESO-1 Cells | Mice NOD/SCID/γnull (NOG) | 10 μM (in vitro) 12.5 mg/kg (in vivo) |
Decreased viability and migration capacity of malignant pleural mesothelioma tumor cells by stimulating autophagy [64]. |
| Lovastatin | Malignant peripheral nerve sheath tumor | NF90-8 and ST88-14 Cells | - | 500 nM | Suppressed viability of cancer cells by inducing non-apoptotic cell death and altering autophagy flux [65]. |
| Lovastatin | Human mesothelioma | Cancer cells ZL55 | - | 2, 8 µM | Reduced the viability of tumor cells by inducing autophagy [66]. |
| Lovastatin | Primary effusion lymphoma (PEL) | BC3 and BCBL1 cells | - | 3, 10, 30 µM | Reduced the survival of PEL cells by triggering apoptotic cell death through the inhibition of autophagic flux [67]. |
| Statin | Cancer Type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Pitavastatin | Pancreatic cancer | ASPC-1 and PANC-1 cells | - | 10 µM | Decreased cell viability by triggering apoptosis, necrosis, and autophagy [68]. |
| Pitavastatin | Melanoma | Human melanoma cells A375 and WM115 | - | 0–5 µM | Induced autophagy and decreased viability of cancer cells [69]. |
| Statin | Cancer type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Rosuvastatin | Papillary thyroid carcinoma | B-CPAP and Nthy-ori 3-1 cells | - | 12,5, 18,5, 25, 50, 100 y 200 µM | Decreased the proliferation and induction of cell death in thyroid cells in a dose- and time-dependent manner [70]. |
| Statin | Cancer type | In Vitro | In Vivo | Dosage | Observation |
|---|---|---|---|---|---|
| Simvastatin | Breast cancer | MDA-MB-231 cells | - | 0.50 µM | Reduced the viability of breast cancer cells by inhibiting autophagy [71]. |
| Simvastatin | Glioma | U251 and C6 cells | - | 0–50 µM | Increased the antiglioma effect through the inhibition of the AMPK-dependent autophagic response [72]. |
| Simvastatin | Brain cancer | GBM cells | - | 0–20 µM | Inhibited temozolomide-induced autophagy flux by blocking autophagolysosome formation [73]. |
References
- Roy, P.S.; Saikia, B. Cancer and cure: A critycal analysis. Indian J. Cancer 2016, 53, 441–442.
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Bray, F. Observatorio Global del Cáncer: Cancer Today; Agencia Internacional de Investigación Sobre el Cáncer: Lyon, France, 2021; Available online: https://gco.iarc.fr/today/home (accessed on 10 December 2021).
- Barbalata, C.I.; Tefas, L.R.; Achim, M.; Tomuta, I.; Porfire, A.S. Statins in risk-reduction and treatment of cancer. World J. Clin. Oncol. 2020, 11, 573–588.
- World Health Organization. Cancer. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 10 December 2021).
- Ho, C.J.; Gorski, S.M. Molecular mechanisms underlying autophagy-mediated treatment resistance in cancer. Cancers 2019, 11, 1775.
- Polivka, J.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175.
- Dey, N.; De, P.; Leyland-Jones, B. Pi3k-AKT-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106.
- Miricescu, D.; Totan, A.; Stanescu-Spinu, L.; Constantin, S.; Constatin, S.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173.
- Afify, S.M.; Oo, A.K.K.; Hassan, G.; Seno, A.; Seno, M. How can we turn the PI3K/AKT/mTOR pathway down? Insights into inhibition and treatment of cáncer. Expert Rev. Anticancer Ther. 2021, 21, 605–619.
- Xu, J.L.; Yuan, L.; Tang, Y.C.; Xu, Z.Y.; Xu, H.D.; Cheng, X.D.; Qin, J.J. The Role of Autophagy in Gastric Cancer Chemoresistance: Friend or Foe? Front. Cell Dev. Biol. 2020, 8, 621428.
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cáncer. Br. J. Cancer 2021, 124, 333–344.
- Botti, J.; Djavaheri-Mergny, M.; Pilatte, Y.; Codogno, P. Autophagy signaling and the cogwheels of cancer. Autophagy 2006, 2, 67–73.
- Chen, N.; Debnath, J. Autophagy and Tumorigenesis. FEBS Lett. 2010, 584, 1427–1435.
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466.
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12.
- Zhang, J.; Yang, Z.; Xie, L.; Xu, L.; Xu, D.; Liu, X. Statins, autophagy and cancer metastasis. Int. J. Biochem. Cell Biol. 2013, 45, 745–752.
- Hassanabad, A.F. Current perspectives on statins as potential anti-cancer therapeutics: Clinical outcomes and underlying molecular mechanisms. Transl. Lung Cancer Res. 2019, 8, 692–699.
- Beckwitt, C.H.; Clark, A.M.; Ma, B.; Whaley, D.; Oltvai, Z.N.; Wells, A. Statins attenuate outgrowth of breast cancer metastases. Br. J. Cancer 2018, 119, 1094–1105.
- Jones, H.M.; Fang, Z.; Sun, W.; Clark, L.H.; Stine, J.E.; Tran, A.Q.; Bae-Jump, V.L. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am. J. Cancer Res. 2017, 7, 2478–2490.
- Chou, C.W.; Lin, C.H.; Hsiao, T.H.; Lo, C.C.; Hsieh, C.Y.; Huang, C.C.; Sher, Y.-P. Therapeutic effects of statins against lung adenocarcinoma via p53 mutant-mediated apoptosis. Sci. Rep. 2019, 9, 1–12.
- Yin, Y.; Liu, L.; Zhao, Z.; Yin, L.; Bauer, N.; Nwaeburu, C.C.; Gladkich, J.; Gross, W.; Hackert, T.; Sticht, C.; et al. Simvastatin inhibits sonic hedgehog signaling and stemness features of pancreatic cancer. Cancer Lett. 2018, 426, 14–24.
- Alipour Talesh, G.; Trezeguet, V.; Merched, A. Hepatocellular Carcinoma and Statins. Biochemistry 2020, 59, 3393–3400.
- Oliveira, K.A.; Dal-Cim, T.; Lopes, F.G.; Ludka, F.K.; Nedel, C.B.; Tasca, C.I. Atorvastatin Promotes Cytotoxicity and Reduces Migration and Proliferation of Human A172 Glioma Cells. Mol. Neurobiol. 2018, 55, 1509–1523.
- Semenkovich Clay, F.; Goldberg Ira, J. 41—Trastornos del metabolismo de los lípidos, Williams. In Tratado de Endocrinología, 14th ed.; Melmed, S., Auchus Richard, J., Goldfine Allison, B., Koenig Ronald, J., Rosen Clifford, J., Eds.; Elsevier: Amsterdam, Neither Land, 2021; pp. 1581–1619. ISBN 978-84-9113-851-8.
- Robinson Jennifer, G. 195—Trastornos del metabolismo de los lípidos, Goldman-Cecil. In Tratado de Medicina Interna, 26th ed.; Goldman, L., Schafer Andrew, I., Eds.; Elsevier: Amsterdam, Neitherland, 2021; pp. 1357–1358. ISBN 978-84-9113-765-8.
- Pedro-Botet Montoya, J.; Masana Marín, L.; Carmena Rodríguez, R. 227—Alteraciones del metabolismo de las lipoproteínas, Farreras Rozman. In Medicina Interna, 19th ed.; von Domarus, A., Farreras, P., Rozman, C., Cardellach, F., Nicolás, J.M., Cervera, R., Agust, A., Brugada, J., Campistol, J.M., et al., Eds.; Elsevier: Amsterdam, Netherlands, 2020; pp. 1813–1837. ISBN 978-84-9113-545-6.
- Fleisher Lee, A.; Beckman Joshua, A. 11—Anestesia y cirugía no cardíaca en pacientes con enfermedades cardíacas, Braunwald. In Tratado de Cardiología, 11th ed.; Zipes Douglas, P., Libby, P., Bonow Robert, O., Mann Douglas, L., Tomaselli Gordon, F., Braunwald, E., Eds.; Elsevier: Amsterdam, Netherlands, 2019; pp. 102–116. ISBN 978-84-9113-398-8.
- Genest, J.; Libby, P. 48—Trastornos de las lipoproteínas y enfermedad cardiovascular, Braunwald. In Tratado de Cardiología, 11th ed.; Zipes Douglas, P., Libby, P., Bonow Robert, O., Mann Douglas, L., Tomaselli Gordon, F., Braunwald, E., Eds.; Elsevier: Amsterdam, Netherlands, 2019; pp. 960–982. ISBN 978-84-9113-398-8.
- Reid Michael, A.; Sanderson Sydney, M.; Locasale Jason, W. 9—Metabolismo del cáncer, Abeloff. In Oncología Clínica, 6th ed.; Niederhuber, J.E., Armitage, J.O., Kastan, M.B., Doroshow, J.H., Tepper, J.E., Eds.; Elsevier: Amsterdam, Netherlands, 2020; pp. 127–138. ISBN 978-84-9113-520-3. Available online: https://www.clinicalkey.es/#!/content/3-s2.0-B9788491135203000096 (accessed on 10 December 2021).
- Araki, M.; Maeda, M.; Motojima, K. Hydrophobic statins induce autophagy and cell death in human rhabdomyosarcoma cells by depleting geranylgeranyl diphosphate. Eur. J. Pharmacol. 2012, 674, 95–103.
- Liao, Y.; Zhang, P.; Yuan, B.; Li, L.; Bao, S. Pravastatin protects against avascular necrosis of femoral head via autophagy. Front. Physiol. 2018, 9, 307.
- Wei, Y.-M.; Li, X.; Xu, M.; Abais, J.M.; Chen, Y.; Riebling, C.R.; Boini, K.M.; Li, P.-L.; Zhang, Y. Enhancement of Autophagy by Simvastatin through Inhibition of Rac1-mTOR Signaling Pathway in Coronary Arterial Myocytes. Cell. Physiol. Biochem. 2013, 31, 925–937.
- Ghavami, S.; Mutawe, M.M.; Sharma, P.; Yeganeh, B.; McNeill, K.D.; Klonisch, T.; Unruh, H.; Kashani, H.; Schaafsma, D.; Los, M.; et al. Mevalonate Cascade Regulation of Airway Mesenchymal Cell Autophagy and Apoptosis: A Dual Role for p53. PLoS ONE 2011, 6, e16523.
- Miettinen, T.P.; Björklund, M. The mevalonate pathway as a metabolic requirement for autophagy–implications for growth control, proteostasis, and disease. Mol. Cell Oncol. 2016, 3, e1143546.
- Zhang, Q.; Yang, Y.-J.; Wang, H.; Dong, Q.-T.; Wang, T.-J.; Qian, H.-Y.; Xu, H. Autophagy Activation: A Novel Mechanism of Atorvastatin to Protect Mesenchymal Stem Cells from Hypoxia and Serum Deprivation via AMP-Activated Protein Kinase/Mammalian Target of Rapamycin Pathway. Stem Cells Dev. 2012, 21, 1321–1332.
- Yang, P.-M.; Chen, C.-C. Life or death? Autophagy in anticancer therapies with statins and histone deacetylase inhibitors. Autophagy 2011, 7, 107–108.
- Tricarico, P.M.; Crovella, S.; Celsi, F. Mevalonate Pathway Blockade, Mitochondrial Dysfunction and Autophagy: A Possible Link. Int. J. Mol. Sci. 2015, 16, 16067–16084.
- Longatti, A.; Tooze, S. Vesicular trafficking and autophagosome formation. Cell Death Differ. 2009, 16, 956–965.
- Chua, C.E.L.; Gan, B.Q.; Tang, B.L. Involvement of members of the Rab family and related small GTPases in autophagosome formation and maturation. Cell Mol. Life Sci. 2011, 68, 3349–3358.
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358.
- Popovic, D.; Akutsu, M.; Novak, I.; Harper, J.W.; Behrends, C.; Dikic, I. Rab GTPase-Activating Proteins in Autophagy: Regulation of Endocytic and Autophagy Pathways by Direct Binding to Human ATG8 Modifiers. Mol. Cell. Biol. 2012, 32, 1733–1744.
- Bodemann, B.O.; Orvedahl, A.; Cheng, T.; Ram, R.R.; Ou, Y.H.; Formstecher, E.; Maiti, M.; .Hazelett, C.C.; Wauson, E.M.; Balakireva, M.; et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 2011, 144, 253–267.
- Sciarretta, S.; Zhai, P.; Shao, D.; Maejima, Y.; Robbins, J.; Volpe, M.; Condorelli, G.; Sadoshima, J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation 2012, 125, 1134–1146.
- Wang, A.-M.; Gao, S.; Zhang, Z.-M.; Shen, Z.-L.; Gao, K.; Chang, L.; Guo, Y.; Li, Z.; Wang, W. Atorvastatin activates autophagy and promotes neurological function recovery after spinal cord injury. Neural Regen. Res. 2016, 11, 977–982.
- Kang, S.Y.; Lee, S.-B.; Kim, H.J.; Yang, H.O.; Jang, W. Autophagic modulation by rosuvastatin prevents rotenone-induced neurotoxicity in an in vitro model of Parkinson’s disease. Neurosci. Lett. 2017, 642, 20–26.
- Vosper, J.; Masuccio, A.; Kullmann, M.; Ploner, C.; Geley, S.; Hengst, L. Statin-induced depletion of geranylgeranyl pyrophosphate inhibits cell proliferation by a novel pathway of Skp2 degradation. Oncotarget 2014, 6, 2889–2902.
- Wang, S.T.; Ho, H.J.; Lin, J.T.; Shieh, J.J.; Wu, C.Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626.
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y.; et al. Fluvastatin Prevents Lung Adenocarcinoma Bone Metastasis by Triggering Autophagy. EBioMedicine 2017, 19, 49–59.
- Ghavami, S.; Yeganeh, B.; Stelmack, G.L.; Kashani, H.H.; Sharma, P.; Cunnington, R.; Freed, D.H. Apoptosis, autophagy and ER stress in mevalonate cascade inhibition-induced cell death of human atrial fibroblasts. Cell Death Dis. 2012, 3, e330.
- Lee, P.S. Simvastatin Induces Mitochondrial Loss and Pten-Mediated Autophagy; San Diego State University: San Diego, CA, USA, 2011; Volume 1, Available online: https://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.991.4451&rep=rep1&type=pdf (accessed on 10 December 2021).
- Zhang, S.-X.; Qiu, L.; Xiao, C.-S.; Fang, Y.-Q. e0034 Inhibition of atorvastatin on the autophagy of vascular endothelial cells. Heart 2010, 96, A11–A12.
- Miettinen, T.P.; Björklund, M. Mevalonate Pathway Regulates Cell Size Homeostasis and Proteostasis through Autophagy. Cell Rep. 2015, 13, 2610–2620.
- Szatmári, Z.; Kis, V.; Lippai, M.; Hegedus, K.; Faragó, T.; Lorincz, P.; Tanaka, T.; Juhász, G.; Sass, M. Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol. Biol. Cell 2014, 25, 522–531.
- Ashrafizadeh, M.; Ahmadi, Z.; Farkhondeh, T.; Samarghandian, S. Modulatory effects of statins on the autophagy: A therapeutic perspective. J. Cell. Physiol. 2019, 235, 3157–3168.
- Yang, P.-M.; Liu, Y.-L.; Lin, Y.-C.; Shun, C.-T.; Wu, M.-S.; Chen, C.-C. Inhibition of Autophagy Enhances Anticancer Effects of Atorvastatin in Digestive Malignancies. Cancer Res. 2010, 70, 7699–7709.
- Parikh, A.; Childress, C.; Deitrick, K.; Lin, Q.; Rukstalis, D.; Yang, W. Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate cancer PC3 cells. Prostate 2010, 70, 971–981.
- Hu, M.-B.; Zhang, J.-W.; Gao, J.-B.; Qi, Y.-W.; Gao, Y.; Xu, L.; Ma, Y.; Wei, Z.-Z. Atorvastatin induces autophagy in MDA-MB-231 breast cancer cells. Ultrastruct. Pathol. 2018, 42, 409–415.
- Alarcon Martinez, T.; Zeybek, N.D.; Müftüoğlu, S. Evaluation of the Cytotoxic and Autophagic Effects of Atorvastatin on MCF-7 Breast Cancer Cells. Balkan Med. J. 2018, 35, 256–262.
- Toepfer, N.; Childress, C.; Parikh, A.; Rukstalis, D.; Yang, W. Atorvastatin induces autophagy in prostate cancer PC3 cells through activation ofLC3transcription. Cancer Biol. Ther. 2011, 12, 691–699.
- Sheng, B.; Song, Y.; Zhang, J.; Li, R.; Wang, Z.; Zhu, X. Atorvastatin suppresses the progression of cervical cancer via regulation of autophagy. Am. J. Transl. Res. 2020, 12, 5252–5268.
- Kang, M.; Jeong, C.W.; Ku, J.H.; Kwak, C.; Kim, H.H. Inhibition of Autophagy Potentiates Atorvastatin-Induced Apoptotic Cell Death in Human Bladder Cancer Cells in Vitro. Int. J. Mol. Sci. 2014, 15, 8106–8121.
- Elimam, H.; El-Say, K.M.; Cybulsky, A.V.; Khalil, H. Regulation of Autophagy Progress via Lysosomal Depletion by Fluvastatin Nanoparticle Treatment in Breast Cancer Cells. ACS Omega 2020, 5, 15476–15486.
- Qi, X.-F.; Kim, D.-H.; Lee, K.-J.; Kim, C.-S.; Song, S.-B.; Cai, D.-Q.; Kim, S.-K. Autophagy contributes to apoptosis in A20 and EL4 lymphoma cells treated with fluvastatin. Cancer Cell Int. 2013, 13, 111.
- Asakura, K.; Izumi, Y.; Yamamoto, M.; Yamauchi, Y.; Kawai, K.; Serizawa, A.; Mizushima, T.; Ohmura, M.; Kawamura, M.; Wakui, M.; et al. The Cytostatic Effects of Lovastatin on ACC-MESO-1 Cells. J. Surg. Res. 2011, 170, e197–e209.
- Wojtkowiak, J.W.; Sane, K.M.; Kleinman, M.; Sloane, B.F.; Reiners, J.J.; Mattingly, R.R. Aborted Autophagy and Nonapoptotic Death Induced by Farnesyl Transferase Inhibitor and Lovastatin. J. Pharmacol. Exp. Ther. 2011, 337, 65–74.
- Shi, Y.; Felley-Bosco, E.; Marti, T.M.; Stahel, R.A. Differential effects of lovastatin on cisplatin responses in normal human mesothelial cells versus cancer cells: Implication for therapy. PLoS ONE 2012, 7, e45354.
- Santarelli, R.; Pompili, C.; Gilardini Montani, M.S.; Romeo, M.A.; Gonnella, R.; D’Orazi, G.; Cirone, M. Lovastatin reduces PEL cell survival by phosphorylating ERK1/2 that blocks the autophagic flux and engages a cross-talk with p53 to activate p21. IUBMB Life 2021, 73, 968–977.
- Chen, Y.; Huang, Y.; Yang, S.; Yen, H.; Tsai, H.; Hsieh, M.; Hsiao, Y. Pitavastatin and metformin synergistically activate apoptosis and autophagy in pancreatic cancer cells. Environ. Toxicol. 2021, 36, 1491–1503.
- Al-Qatati, A.; Aliwaini, S. Combined pitavastatin and dacarbazine treatment activates apoptosis and autophagy resulting in synergistic cytotoxicity in melanoma cells. Oncol. Lett. 2017, 14, 7993–7999.
- Zeybek, N.D.; Gulcelik, N.E.; Kaymaz, F.F.; Sarisozen, C.; Vural, I.; Bodur, E.; Canpinar, H.; Usman, A.; Asan, E. Rosuvastatin induces apoptosis in cultured human papillary thyroid cancer cells. J. Endocrinol. 2011, 210, 105–115.
- Castellanos-Esparza, Y.C.; Wu, S.; Huang, L.; Buquet, C.; Shen, R.; Sanchez-Gonzalez, B.; Latorre, E.A.G.; Boyer, O.; Varin, R.; Jim�Nez-Zamudio, L.; et al. Synergistic promoting effects of pentoxifylline and simvastatin on the apoptosis of triple-negative MDA-MB-231 breast cancer cells. Int. J. Oncol. 2018, 52, 1246–1254.
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V.; et al. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacol. Res. 2012, 65, 111–119.
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034.


