2. Statins
Statins are lipid-lowering drugs that prevent endogenous cholesterol synthesis due to the partial and reversible competitive inhibition of the enzyme hydroxymethyl glutaryl CoA reductase (HMG-CoA reductase), which participates in the mevalonate pathway, an important precursor in the synthesis of cholesterol; however, although this is their main mechanism of action, they have other mechanisms that contribute to the same purpose, such as: increasing LDL receptors, decreasing the release of lipoproteins from the liver to the periphery, increasing the elimination of LDL, reducing the production of lipoproteins, and reducing triglyceride levels by increasing the elimination of VLDL. The pharmacological effects of these mechanisms include reductions in total cholesterol by 20–40% and LDL cholesterol (bad cholesterol) by 20–60%, increases in HDL cholesterol (good cholesterol) of 2–16% and, additionally, decreases in triglycerides of 10–40%. These ranges depend on the active ingredient used, the patient, and the dose prescribed [
152,
153,
154,
155,
156,
157].
It is necessary to highlight that statins have some effects that favor vascular health, such as their anti-inflammatory properties, their reduction of cardiovascular morbidity and mortality in hyperlipidemia, and their ability to induce regression in coronary atherosclerosis and decrease the risk of cerebrovascular accidents [
152,
154,
155]. Statins can be used as preventive or therapeutic measures in all types of hyperlipidemias in which LDL values are increased, such as in hypercholesterolemia and in mixed dyslipidemia; they are very useful for patients with vascular disease and for patients with very high LDL, as well as in moderate cases. In terms of their pharmacokinetics, statins generally require minimal kidney elimination, so they can be used in patients with kidney failure [
152,
157].
Statins can be classified into three groups, according to their efficacy, which depends on the dosage. Some examples of this are rosuvastatin, which can be highly effective (20–40 mg) or moderate (5–10 mg), and atorvastatin (high: 40–80 mg; moderate: 10–20 mg). Other statins that are classified as being moderate and low in efficacy, according to the dose, are: simvastatin, lovastatin, pravastatin, fluvastatin, and pitavastatin [
152].
The side effects of statins that occur most frequently are musculoskeletal symptoms, such as myalgia (5–15%), that are usually symmetrical and located in the legs, where they can occur together with elevated creatine kinase (CK), and gastrointestinal, including abdominal pain, constipation, flatulence, nausea, diarrhea, and headache and fatigue. Another, less frequent, side effect is liver toxicity (2%), which is evidenced by a generally mild increase in hepatic aminotransferases and, therefore, does not require the discontinuation of statins and is dose-dependent; to control this side effect, liver enzyme tests are recommended before starting statin therapy and when it is clinically relevant during treatment, such as to treat the symptoms of hepatitis [
152,
153,
154]. The most potentially serious adverse effect is rhabdomyolysis (infrequent), which generates myoglobinuria and renal failure, which is more frequent in elderly people with generalized weakness, patients with previous renal insufficiency, shock, patients with concomitant disorders, patients who are polymedicated, patients with hypothyroidism, and in perioperative periods [
152,
156]. Typically, it is not necessary to perform routine CK, but it is necessary to request this test in the presence of muscle pain, while the medication is suspended [
154].
Some interactions that can cause side effects of the musculoskeletal type by inhibiting the catabolism of statins, thereby increasing their plasma concentration, occur with drugs that are metabolized in the cytochrome P450 system, such as antifungals (ketoconazole, itraconazole), macrolides (clarithromycin and erythromycin), and other drugs that increase the risk of myopathies, such as fibrates (gemfibrozil), cyclosporine, digoxin (digitalis), calcium channel blockers (verapamil and diltiazem), colchicine, and amiodarone and protease inhibitors. Some factors that also increase the risk of myopathies are grapefruit juice intake, intense physical exercise, and high alcohol intake. The diagnosis of myalgias due to statins is clinical, based on the disappearance of these symptoms when the statin is discontinued and their reappearance when resuming its consumption [
152,
154].
Statins are contraindicated in patients with significant liver dysfunction, in pregnancy, and in ongoing lactation; they can also increase the risk of type 2 diabetes mellitus in patients who have a predisposition to this pathology, as is the case of subjects with metabolic syndrome and prediabetics. This situation has been described when high doses are consumed; therefore, non-pharmacological treatment should be encouraged (diet, exercise), and it is usually continued because the reduction in cardiovascular risk outweighs the possible effect of altering glycemic homeostasis [
152,
153,
154].
Various preclinical studies to date have shown that statins have the ability to effectively induce cell death in different types of human tumor cell. Therefore, statin use is currently an active field of research, within which clinical trials are being developed to establish more clearly whether the use of statins in cancer patients would be beneficial [
157].
2.1. Statins and Autophagy
Evidence has shown that statins can induce autophagy in various types of cell, including vascular endothelial, cardiac, and mesenchymal cells of the respiratory tract, as well as in transformed cells, such as tumor cells [
26,
158,
159,
160]. Statins are a group of drugs that have been used for a long time as cholesterol-lowering agents. They act by inhibiting 3-hydroxy-3-methyl-glutaryl-CoA reductase, a limiting enzyme of the mevalonate pathway. In this pathway, statins also inhibit a variety of other intermediate metabolites, including the formation of isoprenoids, such as farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), leading to the inhibition of the isoprenylation of small GTPases, such as Ras, Rac, Rab, and Rho [
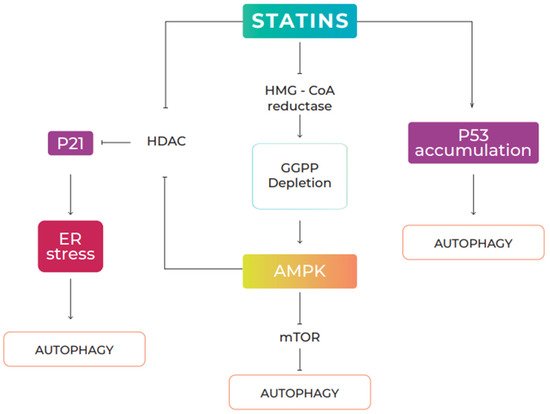
161]. Several signaling pathways have been implicated in the regulation of statin-mediated autophagy, including the AMPK/mTOR (AMP-activated protein kinase/mechanistic target of rapamycin) pathway and the AMPK/p21 pathway. Finally, it has been suggested that p53 accumulation induced by statins can activate autophagy [
160,
162,
163] (
Figure 3).
Figure 3. Statin-induced autophagy signaling pathways. Statins regulate autophagy in several ways: (1) By inhibiting HMG-CoA reductase, statins interfere with the production of mevalonic acid, a precursor in the biosynthesis of geranylgeranyl pyrophosphate (GGPP). Depletion of cellular levels of geranylgeranyl pyrophosphate induces AMPK signaling, repressing mTOR activity, leading to activation of autophagy; (2) statins induce p21 expression through inhibition of histone deacetylase (HDAC) activity, through direct interaction or AMPK-mediated phosphorylation. Activation of the AMPK/p21 signal induced by statins generates ER stress and induces autophagic responses. (3) Statins can increase the accumulation of nuclear p53 and induce autophagy in a p53-dependent manner.
Numerous molecular mediators are involved in autophagy, including the prenylation of GTPase proteins that are indicated as ATG proteins: Rabs, RalB, and Rheb [
164]. Rabs are proteins that act as molecular switches, mediating the transport and fusion of vesicles; they are necessary to develop subdomains in membranes to facilitate maturation [
165]. Recent evidence has shown that some Rabs, including Rab1, Rab5, Rab7, Rab8A, Rab8B, Rab9, Rab11, Rab23, Rab24, Rab25, Rab3, and Rab33B, are essential for autophagy [
166,
167,
168]. The GTPase, RalB, controls crucial physiological processes, including autophagy and invasion. It localizes to a nascent autophagosome and is activated upon nutrient deprivation. Due to its binding to its effector, Exo84, RalB induces the assembly of ULK1 and Beclin1-VPS34, which are necessary for the formation and maturation of autophagosomes [
169]. On the other hand, the brain-enriched Ras homologue (Rheb) regulates cell growth, proliferation, and regeneration through the activation of mTORC1, which results in the inhibition of autophagy by inhibiting the kinase activity of ULK; under starvation conditions, Rheb is inactivated by the Rheb activator protein, GTPase, and mTORC1 is inactivated, leading to the induction of autophagy [
170]. Thus, Rheb inhibition causes statin-induced autophagy.
Another study demonstrated that atorvastatin induced autophagy by enhancing Beclin1 and LC3-II gene expression [
171] and through the AMPK/mTOR pathway [
16]. Similar results were evidenced in endothelial progenitor cells, in which pravastatin enhanced autophagy activity through the upregulation of LC3-II/Beclin-1 and autophagosome formation [
158]; and another study also demonstrated that autophagic modulation by rosuvastatin prevents rotenone-induced neurotoxicity, because rosuvastatin treatment alone increased levels of mTOR-independent/upstream autophagy markers, including Beclin-1 and AMPK [
172]. On the other hand, Wei et al. (2013) observed that simvastatin increased autophagy by inhibiting the Rac1-mTOR pathway [
159]. In primary cultures of human airway smooth muscle (HASM) cells and human atrial fibroblast (hAF) cell lines, statin-induced autophagy and apoptosis were shown to be p53-dependent [
160]. In addition, in vitro tests indicated that statins such as lovastatin and simvastatin caused the degradation of S-phase-associated protein kinase 2 (SKP2) and, consequently, an increase in Beclin1 levels and autophagy [
173,
174].
In two lung adenocarcinoma cell lines, fluvastatin stimulated autophagy activation by increasing LC3-II levels [
175]. Similarly, Ghavami et al. (2012) showed that statins induce apoptosis and autophagy in the mesenchymal cells of the human lung, and also suggested that autophagy has a crucial role in determining the degree of stress of the endoplasmic reticulum (ER), unfolded protein response (UPR), and cell-line permissiveness (hAF) to statin-induced death [
176]. Another study indicated that simvastatin induced autophagy in cardiac cell lines and in mouse hearts in vivo; these cells were treated with simvastatin, producing a slight mitochondrial depolarization relative to controls and a significant increase in PTEN levels. This shows that simvastatin is capable of inducing PTEN-mediated mitochondrial autophagy, which is related to its cardioprotective capacity [
177].
Some studies have shown that statins can inhibit autophagy. For example, atorvastatin inhibits vascular endothelial cell autophagy, an effect that may be related to atorvastatin’s role in improving endothelial function. However, the use of atorvastatin, before the onset of induced autophagy, cannot effectively inhibit autophagy [
178]. Another study showed that the inhibition of the mevalonate pathway simultaneously blocks autophagosome maturation, leading to reduced autophagic flux [
179]. Similarly, one study reported that autophagic flux block is caused by reduced prenylation and, more specifically, the geranylgeranylation of Rab11, a small GTPase required for autophagosome formation, which acts in the recycling endosome [
180]. Thus, mevalonate pathway activity functions as a metabolic requirement for basal autophagic flux through Rab11 geranylgeranylation.
Taken together, these studies demonstrate that the role of statins in autophagy is related to the expression levels of GGPP and prenylated proteins, making them important players in this mechanism. Therefore, the therapeutic effects of statins in various cell types have been attributed to the modulation of autophagy, which is essential for maintaining cellular homeostasis and explains the elimination of unfavorable cells or specific organelles within cells.
2.2. Effects of Statins on Cancer via Autophagy
Statins have emerged as a potential therapy against cancer due to their effects on autophagy [
181]. Several studies have shown that the role of autophagy in tumorigenesis is very contradictory, since its induction could accelerate tumor progression or, on the contrary, induce tumor suppression [
175]. In accordance with the important role that autophagy plays in the pathological conditions of cancer, compounds with proautophagic or antiautophagic modulating effects are of pharmacological interest [
181].
In preclinical studies, statins have been shown to be capable of inducing autophagy in some cancer cells by inhibiting geranylgeranyl [
26,
182,
183]. The antitumor activity of atorvastatin (ATO) has been shown to be associated with the induction of autophagy in breast cancer cells [
25,
184] and ovarian cancer [
19]. ATO induces autophagy in PC3 prostate cancer cells by activating LC3 transcription [
185]. Sheng et al. (2020) showed that ATO inhibited tumor growth and promoted the apoptosis of cervical cancer both in vitro and in vivo, which could be associated with the suppression of the mevalonate pathway. Furthermore, ATO could also induce autophagy in cervical cancer cells, but, more importantly, the pharmacological inhibition of autophagy significantly enhanced ATO-induced cytotoxicity in cervical cancer [
24]. Similarly, Yan et al. (2014) showed that ATO induces autophagy in Huh7 and HCT116 gastrointestinal tumor cell lines and that combinations of ATO with autophagy inhibitors provide a novel and promising strategy to improve the treatment of digestive malignancies [
182]. Another study suggests that the inhibition of autophagy potentiates ATO-induced apoptotic cell death in human bladder cancer cells [
186] (
Table 1).
Table 1. In vitro and in vivo studies on the anticancer potential of atorvastatin via autophagy.
| Statin |
Cancer Type |
In Vitro |
In Vivo |
Dosage |
Observation |
| Atorvastatin |
Breast cancer |
MDA-MB-231 Cells |
- |
0,5, 1, 2, 4, 8 µM |
Reduced the viability of cancer cells by inducing autophagy [25]. |
| Atorvastatin |
Breast cancer |
MCF-7 |
- |
5, 10, 20, 40 y 80 μM |
Decreased the proliferation of breast cancer cells through the induction of both apoptosis and autophagy [184]. |
| Atorvastatin |
Ovarian cancer |
Hey and SKOV3 cells |
- |
1–250 μM |
Inhibited the growth of ovarian cancer cell lines associated with the induction of apoptosis, autophagy, cellular stress, and G1 cell-cycle arrest [19]. |
| Atorvastatin |
Cervical Cancer |
SiHa and Caski Cells |
Female BALB/c
nude mice |
0, 5, 10, y 20, 40, 80 μM (in vitro)
50 mg/kg (in vivo) |
Reduced the viability of cervical cancer cells in vitro and in vivo by inducing apoptosis. ATO induced autophagy, and its inhibition was shown to enhance the anti-cancer effects of ATO on cervical cancer cells [24]. |
| Atorvastatin |
Digestive malignancies |
HCC cells (Hep3B, HepG2 and Huh7)
CRC cells (HCT116 wt, HCT116 p21) |
Female BALB
nude mice |
50 μM
(in vitro)
50 mg/kg (in vivo) |
Inhibited cancer cell growth in vivo and in vitro by inducing apoptosis. ATO induced autophagy, and the pharmacological inhibition of autophagy was shown to enhance the anticancer effects of ATO in gastrointestinal malignancies [182]. |
| Atorvastatin |
Bladder Cancer |
T24 and J28 Cells |
- |
0, 10, 20, 30, 40 y 50 μM |
Enhanced ATP-induced apoptotic cell death in human bladder cancer cells in vitro through the pharmacological inhibition of autophagy [186]. |
Fluvastatin has been reported to induce autophagy in breast cancer cells through AMPK phosphorylation and the inhibition of basal extracellular signal-regulated kinase (ERK) phosphorylation [
187]. Fluvastatin can suppress the bone metastasis of lung adenocarcinoma by inducing nuclear wild-type p53 expression, activating AMPK-mTOR-dependent autophagy in cancer cells [
175]. Fluvastatin induced apoptosis in lymphoma cells by regulating autophagy through the inhibition of metabolic products of the HMG-CoA reductase reaction [
188] (
Table 2). Asakura et al. (2011) studied the antitumor activity of lovastatin in malignant pleural mesothelioma cells. Their results demonstrated that lovastatin administration reduced primary tumors and metastasis in a NOG mouse model of human malignant mesothelioma. In vitro studies demonstrated that lovastatin administration induced cytostatic effects, such as reduced cell viability and cell migration, in ACC-MESO-1 cells. The authors suggested that these effects were dependent on autophagic changes rather than apoptosis; furthermore, the induction of autophagic changes by lovastatin in ACC-MESO-1 cells was independent of mTOR and was considered to be dependent, at least in part, on Rac/phospholipase C/inositol 1,4,5-triphosphate [
189]. Wojtkowiak et al. (2011) found that the combination of lovastatin and a farnesyl transferase inhibitor (FTI-1) induced a dysfunctional autophagic program and non-apoptotic cell death in malignant liver sheath tumor cell lines in human peripheral nerves [
190]. Another study showed that the combination of lovastatin and cisplatin improves LC3B-II levels and reduces the viability of cancer cells by inducing autophagic cell death [
191]. On the other hand, a recent study indicated that lovastatin decreases the survival of PEL cells, an aggressive B-cell lymphoma, by phosphorylating ERK1/2, blocking autophagic flux [
192] (
Table 3).
Table 2. In vitro and in vivo studies on the anticancer potential of fluvastatin via autophagy.
| Statin |
Cancer Type |
In Vitro |
In Vivo |
Dosage |
Observation |
| Fluvastatin |
Breast cancer |
MCF-7 |
- |
10 μM |
Reduced cell viability through the depletion of lysosomal activities coupled with the accumulation of autophagosomes, leading to impaired autophagosome–lysosomal fusion in treated cells [187]. |
| Fluvastatin |
Lung adenocarcinoma |
A549 and SPC-A-1 cells |
Female nude mice BALB/c |
10 μM (in vitro)
50 mg/kg (in vivo) |
Suppressed bone metastasis from lung adenocarcinoma in vivo and in vitro by triggering autophagy through the p53–AMPK-mTOR pathway [175]. |
| Fluvastatin |
Lymphoma |
A20 and EL4 cells |
- |
0–10 μM |
Induced apoptosis in lymphoma cells by activating autophagy through increased LC3-II [188]. |
Table 3. In vitro and in vivo studies on the anticancer potential of lovastatin via autophagy.
| Statin |
Cancer Type |
In Vitro |
In Vivo |
Dosage |
Observation |
| Lovastatin |
Malignant pleural mesothelioma |
ACC-MESO-1 Cells |
Mice NOD/SCID/γnull (NOG) |
10 μM (in vitro)
12.5 mg/kg (in vivo) |
Decreased viability and migration capacity of malignant pleural mesothelioma tumor cells by stimulating autophagy [189]. |
| Lovastatin |
Malignant peripheral nerve sheath tumor |
NF90-8 and ST88-14 Cells |
- |
500 nM |
Suppressed viability of cancer cells by inducing non-apoptotic cell death and altering autophagy flux [190]. |
| Lovastatin |
Human mesothelioma |
Cancer cells ZL55 |
- |
2, 8 µM |
Reduced the viability of tumor cells by inducing autophagy [191]. |
| Lovastatin |
Primary effusion lymphoma (PEL) |
BC3 and BCBL1 cells |
- |
3, 10, 30 µM |
Reduced the survival of PEL cells by triggering apoptotic cell death through the inhibition of autophagic flux [192]. |
Researchers have shown that pitavastatin, another class of statins, in conjunction with metformin could preserve mitochondrial function, activate AMPK, and inhibit PI3K/mTOR, unlike treatment with metformin or pitavastatin alone. These findings clearly indicated that metformin plus pitavastatin had a synergistic anticancer effect on pancreatic cancer cells, which was potentially caused by the activation of AMPK and the inhibition of PI3K/mTOR signaling [
193]. The antitumor activity of pitavastatin was also investigated in melanoma cells; the results demonstrated that melanoma cells treated with combined pitavastatin–dacarbazine (DTIC) expressed a high level of LC3-II, a marker of autophagy. The chemical inhibition of autophagy resulted in enhanced cell viability, suggesting that pitavastatin–DTIC-induced autophagy occurs as a mechanism of cell death.
In support of this, it has been suggested that DTIC and pitavastatin may have a role in the induction of autophagy, specifically as a mode of cell death. Therefore, the pitavastatin–DTIC combination treatment provides a synergistic anticancer effect through apoptosis and autophagy [
194] (
Table 4). On the other hand, a study showed that treatment with rosuvastatin can be an alternative for patients with papillary thyroid cancer. Rosuvastatin induced autophagic changes in papillary thyroid carcinoma (B-CPAP) cells, even at low doses, showing a variation from autophagic changes to apoptosis with increasing concentrations of rosuvastatin. In normal thyroid cells (Nthy-ori3-1), however, minimal/early autophagic changes were observed only at higher doses and with increased exposure times. The results indicated that rosuvastatin treatment induced autophagy and subsequent apoptosis in B-CPAP cells [
195] (
Table 5).
Table 4. In vitro and in vivo studies on the anticancer potential of pitavastatin via autophagy.
| Statin |
Cancer Type |
In Vitro |
In Vivo |
Dosage |
Observation |
| Pitavastatin |
Pancreatic cancer |
ASPC-1 and PANC-1 cells |
- |
10 µM |
Decreased cell viability by triggering apoptosis, necrosis, and autophagy [193]. |
| Pitavastatin |
Melanoma |
Human melanoma cells A375 and WM115 |
- |
0–5 µM |
Induced autophagy and decreased viability of cancer cells [194]. |
Table 5. In vitro and in vivo studies on the anticancer potential of rosuvastatin via autophagy.
| Statin |
Cancer type |
In Vitro |
In Vivo |
Dosage |
Observation |
| Rosuvastatin |
Papillary thyroid carcinoma |
B-CPAP and Nthy-ori 3-1 cells |
- |
12,5, 18,5, 25, 50, 100 y 200 µM |
Decreased the proliferation and induction of cell death in thyroid cells in a dose- and time-dependent manner [195]. |
Castellanos et al. (2018) have shown that simvastatin (SIM) mainly induces cell apoptosis, while pentoxifylline (PTX) induces autophagy and cell apoptosis; the two drugs used in combination reduced autophagy rates while apoptosis levels increased inversely. These results strongly support the existence of a well-regulated balance between apoptosis and autophagy. Thus, the study most likely revealed an interesting pharmaceutical property of simvastatin, indicating that it is capable of disrupting equilibrium, altering cell fate, and inducing cell death by stimulating extracellular regulated protein kinases 1 and 2 (ERK1/2) and Akt pathways. The results therefore suggest that the induction of autophagy may be a protective mechanism preventing the death of MDA-MB-231 breast cancer cells and that the combined use of PTX and SIM may render quiescent autophagic cancer cells experiencing apoptosis. Therefore, this may be a new treatment strategy for triple-negative breast cancer [
196].
The role of autophagy in glioma cell death induced by the drug simvastatin has been investigated, suggesting that the inhibition of the AMPK-dependent autophagic response could sensitize glioma cells to apoptotic death induced by simvastatin [
197]. The anticancer effects of the chemotherapeutic agent temozolomide (TMZ), a drug used for the treatment of glioblastoma (GBM), is significantly reduced by autophagy induced by TMZ. One study demonstrated that simvastatin inhibits TMZ-induced autophagic flux by blocking autophagosome–lysosome formation, thus sensitizing glioblastoma cells to TMZ-induced cell death, making it a promising therapeutic strategy for GBM treatment [
198] (
Table 6).
Table 6. In vitro and in vivo studies on the anticancer potential of simvastatin via autophagy.
| Statin |
Cancer type |
In Vitro |
In Vivo |
Dosage |
Observation |
| Simvastatin |
Breast cancer |
MDA-MB-231 cells |
- |
0.50 µM |
Reduced the viability of breast cancer cells by inhibiting autophagy [196]. |
| Simvastatin |
Glioma |
U251 and C6 cells |
- |
0–50 µM |
Increased the antiglioma effect through the inhibition of the AMPK-dependent autophagic response [197]. |
| Simvastatin |
Brain cancer |
GBM cells |
- |
0–20 µM |
Inhibited temozolomide-induced autophagy flux by blocking autophagolysosome formation [198]. |
In vitro and in vivo investigations indicate that a number of beneficial effects of statins are due to mechanisms that predominantly interfere with tumor growth and invasion. Autophagy, a self-degradation pathway, acts as a double-edged sword in cancer. Statins can induce autophagy in some cancer cells. Statins have been shown to inhibit the cell viability of multiple cancers, including ovarian cancer, lung adenocarcinoma, malignant pleural mesothelioma, melanoma, and pancreatic cancer by inducing autophagy. The combination of statins with autophagic inhibitors is a new and promising strategy to improve the treatment of digestive malignancies, cervical cancer, and bladder cancer. This means that autophagy plays a fundamental role in pathological conditions. However, more studies are needed in the future to fully understand the modulatory effect of statins on autophagy.